
Tramadol
- CAS NO.:27203-92-5
- Empirical Formula: C16H25NO2
- Molecular Weight: 263.38
- MDL number: MFCD00941467
- EINECS: 248-319-6
- SAFETY DATA SHEET (SDS)
- Update Date: 2024-12-18 14:08:57
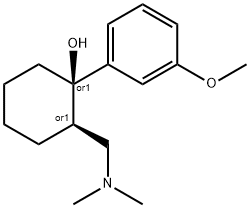
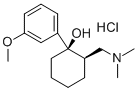
What is Tramadol?
Absorption
Oral Administration
Tramadol is administered as a racemate, with both the [-] and [+] forms of both tramadol and the M1 metabolite detected in circulation. Following administration, racemic tramadol is rapidly and almost completely absorbed, with a bioavailability of 75%. This difference in absorption and bioavailability can be attributed to the 20-30% first-pass metabolism. Peak plasma concentrations of tramadol and the primary metabolite M1 occur at two and three hours, respectively. Following a single oral dose of 100mg of tramadol, the Cmax was found to be approximately 300μg/L with a Tmax of 1.6-1.9 hours, while metabolite M1 was found to have a Cmax of 55μg/L with a Tmax of 3 hours.
Steady-state plasma concentrations of both tramadol and M1 are achieved within two days of dosing. There is no evidence of self-induction. Following multiple oral doses, Cmax is 16% higher and AUC is 36% higher than after a single dose, demonstrating a potential role of saturable first-pass hepatic metabolism in increasing bioavailability.
Intramuscular Administration
Tramadol is rapidly and almost completely absorbed following intramuscular administration. Following injection of 50mg of tramadol, Cmax of 166μg/L was found with a Tmax of 0.75 hours.
Rectal Administration
Following rectal administration with suppositories containing 100mg of tramadol, Cmax of 294μg/L was found with a Tmax of 3.3 hours. The absolute bioavailability was found to be higher than oral administration (77% vs 75%), likely due to reduced first-pass metabolism with rectal administration compared to oral administration.
Toxicity
The reported LD50 for tramadol, when administered orally in mice, is 350 mg/kg.
In carcinogenic studies, there are reports of murine tumors which cannot be concluded to be carcinogenic in humans. On the other hand, tramadol showed no evidence to be mutagenic in different assays and does not have effects on fertility. However, there are clear reports of embryotoxicity and fetotoxicity.
Description
Tramadol is a venerable opioid analgesic. Its preparation was first reported in 1965 in a British patent to the West German company Gr?nenthal GmbH. It was not until 1977 that Gr?nenthal marketed the drug under the trade name Tramal. Twenty years later, companies in the United States, the United Kingdom, and Australia began to sell tramadol under various names.
Commercial tramadol consists?primarily of two of its four stereoisomers, (R,R) and (S,S). (R,R)-tramadol is shown in the images. It has at least six known mechanisms of action, including as a μ-opioid receptor agonist and as a serotonin releasing agent. It has serious side effects, including seizures and risk of suicide.
Tramadol is a synthetic product that was designed to contain the structural portion of morphine that gives it its pain-killing activity. But in 2013, M. De Waard and several colleagues in France, Cameroon, and Switzerland?reported its occurrence in the Cameroonian pincushion tree?(Nauclea latifolia). Although De Waard’s experiments took pains to rule out contamination of the plants by synthetic tramadol, in September 2014, however, a study by M. Spiteller and coauthors in Germany and Cameroon showed that tramadol in the trees is the result of human contamination, most likely because natives in northern Cameroon administer the drug to working farm animals so that they tire less easily. The authors believe that the animals’ urine contaminates the soil where the trees grow.
Two other scientists in the field are not convinced by either research team’s argument. All of the parties are conducting additional studies to resolve the question. The answer may depend on whether the isotope ratios and stereoisomer distribution of synthetic and “natural” tramadol match up.
Chemical properties
Light Yellow Oil
Originator
Tramadol,Gruenenthal,W. Germany,1977
The Uses of Tramadol
Tramadol is thought to produce analgesia by two distinct actions. First, it has
agonist activity at the MOP and KOP receptors. Tramadol itself is a prodrug,
with most of its analgesia mediated by a metabolite – O-desmethyltramadol –
that has a 200-fold higher affinity for the MOP receptor. I t is metabolised by
cytochrome P450 (CYP2D6 and CYP3A4), and its potency is therefore
affected by a patient's CYP genetics, with rapid and poor metabolisers.
S econd, it enhances the descending inhibitory systems in the spinal cord by inhibiting noradrenaline reuptake and releasing serotonin from nerve
endings. It is available in immediate- and sustained-release oral preparations
and for parenteral administration. I ts use is contraindicated in patients
receiving monoamine oxidase inhibitors (MAOIs). Caution must also be
exercised in hepatic impairment as its clearance is reduced to a much greater
extent than morphine and related agents.
The Uses of Tramadol
An Analgesic. Used in the treatment of urinary incontinence
Background
Tramadol is a centrally acting synthetic opioid analgesic and SNRI (serotonin/norepinephrine reuptake-inhibitor) that is structurally related to codeine and morphine. Due to its good tolerability profile and multimodal mechanism of action, tramadol is generally considered a lower-risk opioid option for the treatment of moderate to severe pain. It is considered a Step 2 option on the World Health Organization's pain ladder and has about 1/10th of the potency of morphine.
Tramadol differs from other traditional opioid medications in that it doesn't just act as a μ-opioid agonist, but also affects monoamines by modulating the effects of neurotransmitters involved in the modulation of pain such as serotonin and norepinpehrine which activate descending pain inhibitory pathways. Tramadol's effects on serotonin and norepinephrine mimic the effects of other SNRI antidepressants such as duloxetine and venlafaxine.
Tramadol exists as a racemic mixture consisting of two pharmacologically active enantiomers that both contribute to its analgesic property through different mechanisms and are also themselves metabolized into active metabolites: (+)-tramadol and its primary metabolite (+)-O-desmethyl-tramadol (M1) are agonists of the μ opioid receptor while (+)-tramadol inhibits serotonin reuptake and (-)-tramadol inhibits norepinephrine reuptake. These pathways are complementary and synergistic, improving tramadol's ability to modulate the perception of and response to pain.
Tramadol has also been shown to affect a number of other pain modulators within the central nervous system as well as non-neuronal inflammatory markers and immune mediators. Due to the broad spectrum of targets involved in pain and inflammation, it's not surprising that the evidence has shown that tramadol is effective for a number of pain types including neuropathic pain, post-operative pain, lower back pain, as well as pain associated with labour, osteoarthritis, fibromyalgia, and cancer. Due to its SNRI activity, tramadol also has anxiolytic, antidepressant, and anti-shivering effects which are all frequently found as comorbidities with pain.
Similar to other opioid medications, tramadol poses a risk for development of tolerance, dependence and abuse. If used in higher doses, or with other opioids, there is a dose-related risk of overdose, respiratory depression, and death. However, unlike other opioid medications, tramadol use also carries a risk of seizure and serotonin syndrome, particularly if used with other serotonergic medications.
Indications
Tramadol is approved for the management of moderate to severe pain in adults.
Tramadol is also used off-label in the treatment of premature ejaculation.
Definition
ChEBI: (R,R)-tramadol is a 2-[(dimethylamino)methyl]-1-(3-methoxyphenyl)cyclohexanol in which both stereocentres have R-configuration; the (R,R)-enantiomer of the racemic opioid analgesic tramadol, it exhibits ten-fold higher analgesic potency than the (S,S)-enantiomer. It has a role as a delta-opioid receptor agonist, a kappa-opioid receptor agonist, a mu-opioid receptor agonist, an adrenergic uptake inhibitor, an antitussive, a capsaicin receptor antagonist, a muscarinic antagonist, a nicotinic antagonist, a NMDA receptor antagonist, an opioid analgesic, a serotonergic antagonist, a serotonin uptake inhibitor and a metabolite. It is a conjugate base of a (R,R)-tramadol(1+). It is an enantiomer of a (S,S)-tramadol.
Manufacturing Process
5 g of magnesium turnings are treated while stirring with a mixture of 37.4 g
of m-bromoanisole and 160 ml of absolute tetrahydrofuran at such a rate that
the reaction mixture boils gently because of the heat produced by the
immediately starting reaction. Thereafter, the reaction mixture is boiled under
reflux while stirring until all the magnesium dissolves. The reaction mixture is
cooled to 0°C to -10°C and then a mixture of 23.25 g of 2-
dimethylaminomethylcyclohexanone and 45 ml of absolute tetrahydrofuran is
added dropwise.
The resulting mixture is stirred for 4 hours at room temperature and then
poured, while stirring slowly, into a mixture of 25 g of ammonium chloride, 50
ml of water and 50 g of ice. The layers are separated and the aqueous layer is
extracted twice with 50 ml portions of ether. The organic layers are combined,
dried with sodium sulfate and evaporated. The residue is distilled, and 1-(m_x0002_methoxyphenyl)-2-dimethylaminomethyl-cyclohexanol-(1), boiling point at 0.6
mm Hg 138°C to 140°C, is obtained in a yield of 78.6% of theoretical. The hydrochloride obtained from the product, e.g., by dissolving in ether and
treating with dry hydrogen chloride, melts at 168°C to 175°C. By
recrystallization from moist dioxan this hydrochloride is separated into isomers
melting at 162°C to 163°C and 175°C to 177°C, respectively.
brand name
Trabar.
Therapeutic Function
Analgesic
General Description
Tramadol (Ultram) is an analgesic agent with multiple mechanismsof action. It is a weak μ-agonist with approximately30% of the analgesic effect antagonized by the opioid antagonistnaloxone. Used at recommended doses, it has minimaleffects on respiratory rate, heart rate, blood pressure, or GItransit times. Structurally, tramadol resembles codeine with the B, D, and E ring removed. The manufacturer states thatpatients allergic to codeine should not receive tramadol, becausethey may be at increased risk for anaphylactic reactions. Tramadol is synthesized and marketed as the racemicmixture of two (the [2S, 3S] [-] and the [2R, 3R] [+]) of thefour possible enantiomers. The (+) enantiomer is about30 times more potent than the (—) enantiomer; however,racemic tramadol shows improved tolerability.Neurotransmitter reuptake inhibition is also responsible forsome of the analgesic activity with the (—) enantiomer primarilyresponsible for norepinephrine reuptake and the (+)enantiomer responsible for inhibiting serotonin reuptake. Like codeine, tramadol is O-demethylated viaCYP2D6 to a more potent opioid agonist having 200-foldhigher affinity for the opioid receptor than the parent compound.Tramadol was initially marketed as nonaddictive, anda 3-year follow up study showed that the abuse potential isvery low, but not zero. Most abusers of tramadol have abusedopioid drugs in the past. Both enantiomers of tramadoland the major O-demethylated metabolite are proconvulsive,and tramadol should not be used in patients with a lowseizurethreshold including patients with epilepsy.
Mechanism of action
Fentanyl is a μ agonist with approximately 80 times greater potency than morphine. Fentanyl has been used in combination with nitrous oxide for “ balanced” anesthesia and in combination with droperidol for “ neurolepalgesia.” The advantages of fentanyl over morphine for anesthetic procedures are its shorter duration of action (1–2 hours) and the fact that it does not cause histamine release on intravenous injection.
Pharmacokinetics
Tramadol modulates the descending pain pathways within the central nervous system through the binding of parent and M1 metabolite to μ-opioid receptors and the weak inhibition of the reuptake of norepinephrine and serotonin.
Apart from analgesia, tramadol may produce a constellation of symptoms (including dizziness, somnolence, nausea, constipation, sweating and pruritus) similar to that of other opioids.
Central Nervous System
In contrast to morphine, tramadol has not been shown to cause histamine release. At therapeutic doses, tramadol has no effect on heart rate, left-ventricular function or cardiac index. Orthostatic hypotension has been observed.
Tramadol produces respiratory depression by direct action on brain stem respiratory centres. The respiratory depression involves both a reduction in the responsiveness of the brain stem centres to increases in CO2 tension and to electrical stimulation.
Tramadol depresses the cough reflex by a direct effect on the cough centre in the medulla. Antitussive effects may occur with doses lower than those usually required for analgesia.
Tramadol causes miosis, even in total darkness. Pinpoint pupils are a sign of opioid overdose but
are not pathognomonic (e.g., pontine lesions of hemorrhagic or ischemic origin may produce
similar findings). Marked mydriasis rather than miosis may be seen with hypoxia in the setting of
oxycodone overdose.
Seizures have been reported in patients receiving tramadol within the recommended dosage range. Spontaneous post-marketing reports indicate that seizure risk is increased with doses of tramadol above the recommended range. Risk of convulsions may also increase in patients with epilepsy, those with a history of seizures
or in patients with a recognized risk for seizure (such as head trauma, metabolic disorders,
alcohol and drug withdrawal, CNS infections), or with concomitant use of other drugs known to reduce the seizure threshold.
Tramadol can cause a rare but potentially life-threatening condition resulting from concomitant administration of serotonergic drugs (e.g., anti-depressants, migraine medications). Treatment with the serotoninergic drug should be discontinued if such events (characterized by clusters of symptoms such as hyperthermia, rigidity, myoclonus, autonomic instability with possible rapid fluctuations of vital signs, mental status changes including confusion, irritability, extreme agitation progressing to delirium and coma) occur and supportive symptomatic treatment should be initiated. Tramadol should not be used in combination with MAO inhibitors or serotonin-precursors (such as L-tryptophan, oxitriptan) and should be used with caution in combination with other serotonergic drugs (triptans, certain tricyclic antidepressants, lithium, St. John’s Wort) due to the risk of serotonin syndrome.
Gastrointestinal Tract and Other Smooth Muscle
Tramadol causes a reduction in motility associated with an increase in smooth muscle tone in the antrum of the stomach and duodenum. Digestion of food in the small intestine is delayed and propulsive contractions are decreased. Propulsive peristaltic waves in the colon are decreased, while tone may be increased to the point of spasm resulting in constipation. Other opioid-induced effects may include a reduction in gastric, biliary and pancreatic secretions, spasm of the sphincter of Oddi, and transient elevations in serum amylase.
Endocrine System
Opioids may influence the hypothalamic-pituitary-adrenal or -gonadal axes. Some changes that can be seen include an increase in serum prolactin and decreases in plasma cortisol and testosterone. Clinical signs and symptoms may be manifest from these hormonal changes.
Hyponatremia has been reported very rarely with the use of tramadol, usually in patients with predisposing risk factors, such as elderly patients and/or patients using concomitant medications that may cause hyponatremia (e.g., antidepressants, benzodiazepines, diuretics). In some reports, hyponatremia appeared to be the result of the syndrome of inappropriate antidiuretic hormone secretion (SIADH) and resolved with discontinuation of tramadol and appropriate treatment (e.g., fluid restriction). During tramadol treatment, monitoring for signs and symptoms of hyponatremia is recommended for patients with predisposing risk factors.
Cardiovascular
Tramadol administration may result in severe hypotension in patients whose ability to maintain adequate blood pressure is compromised by reduced blood volume, or concurrent administration of drugs such as phenothiazines and other tranquillizers, sedative/hypnotics, tricyclic antidepressants or general anesthetics. These patients should be monitored for signs of hypotension after initiating or titrating the dose of tramadol.
QTc-Interval Prolongation
The maximum placebo-adjusted mean change from baseline in the QTcF interval was 5.5 ms in the 400 mg/day treatment arm and 6.5 ms in the 600 mg/day mg treatment arm, both occurring at the 8h time point. Both treatment groups were within the 10 ms threshold for QT prolongation. Post-marketing experience with the use of tramadol containing products included rare reports of QT prolongation reported with an overdose. Particular care should be exercised when administering tramadol to patients who are suspected to be at an increased risk of experiencing torsade de pointes during treatment with a QTc-prolonging drug.
Abuse and Misuse
Like all opioids, tramadol has the potential for abuse and misuse, which can lead to overdose and death. Therefore, tramadol should be prescribed and handled with caution.
Dependence/Tolerance
Physical dependence and tolerance reflect the neuroadaptation of the opioid receptors to chronic exposure to an opioid and are separate and distinct from abuse and addiction. Tolerance, as well as physical dependence, may develop upon repeated administration of opioids, and are not by themselves evidence of an addictive disorder or abuse. Patients on prolonged therapy should be tapered gradually from the drug if it is no longer required for pain control. Withdrawal symptoms may occur following abrupt discontinuation of therapy or upon administration of an opioid antagonist. Some of the symptoms that may be associated with abrupt withdrawal of an opioid analgesic include body aches, diarrhea, gooseflesh, loss of appetite, nausea, nervousness or restlessness, anxiety, runny nose, sneezing, tremors or shivering, stomach cramps, tachycardia, trouble with sleeping, unusual increase in sweating, palpitations, unexplained fever, weakness and yawning.
Pharmacokinetics
The analgesic activity of tramadol is attributed to a synergistic effect caused by the opioid activity
of the (+)-isomer and the neurotransmitter reuptake blocking effect of the (–)-isomer. The
(+)-isomer possesses weak μ opioid agonist activity equivalent to approximately 1/3,800 that of
morphine. The O-desmethyl metabolite (CYP2D6) of (±)-tramadol has improved μ opioid activity
equivalent to 1/35 that of morphine. Affinity for both δ and κ receptors is improved. Despite its
higher opioid potency, the contribution of O-desmethlytramedol to the overall analgesic effect has
been questioned but not well studied. Individuals who lack CYP2D6 or are taking a CYP2D6
inhibitor have a reduced effect to tramadol. The fact that naloxone causes a decrease in the
analgesic potency of tramadol argues strongly for an opioid component to the analgesic activity.
(–)-T ramadol possesses only 1/20 the opioid activity of its (+)-isomer, but it has good activities for
inhibition of norepinephrine (Ki = 0.78 μM) and serotonin (Ki = 0.99 μM) reuptake. Tramadol's
neurotransmitter reuptake activity is approximately 1/20 that of imipramine, a tricyclic
antidepressant agent that is used widely in pain management. Although none of the individual
pharmacological activities of tramadol is impressive, they interact to give a synergistic analgesic
effect that is clinically useful.
Tramadol has been used in Europe since the 1980s and was introduced to the U.S. market in 1995.
The drug is nonaddicting and, thus, is not a scheduled agent. In addition, tramadol does not cause
respiratory depression or constipation.
Metabolism
Tramadol undergoes extensive first-pass metabolism in the liver by N- and O- demethylation and conjugation. From the extensive metabolism, there have been identified at least 23 metabolites. There are two main metabolic pathways: the O-demethylation of tramadol to produce O-desmethyl-tramadol (M1) catalyzed by CYP2D6 and the N-demethylation to N-desmethyl-tramadol (M2) catalyzed by CYP3A4 and CYP2B6.
The wide variability in the pharmacokinetic properties between patients can partly be ascribed to polymorphisms within the gene for CYP2D6 that determine its enzymatic activity. CYP2D6*1 is considered the wild-type allele associated with normal enzyme activity and the "extensive metabolizer" phenotype; 90-95% of Caucasians are considered "extensive metabolizers" (with normal CYP2D6 function) while the remaining 5-10% are considered "poor metabolizers" with reduced or non-functioning enzyme. CYP2D6 alleles associated with non-functioning enzyme include *3, *4, *5, and *6 while alleles associated with reduced activity include *9, *10, *17, and *41.
Poor metabolizers have reduced activity of the CYP2D6 enzyme and therefore less production of tramadol metabolites M1 and M2, which ultimately results in a reduced analgesic effect as tramadol interacts with the μ-opioid receptor primarily via M1.
There are also large differences in the frequency of these alleles between different ethnicities: *3, *4, *5, *6, and *41 are more common among Caucasians while *17 is more common in Africans for example. Compared to 5-10% of Caucasians, only ~1% of Asians are considered poor metabolizers, however Asian populations carry a much higher frequency (51%) of the CYP2D6*10 allele, which is relatively rare in Caucasian populations and results in higher exposure to tramadol.
Some individuals are considered "ultra-rapid metabolizers", such as those carrying CYP2D6 gene duplications (CYP2D6*DUP) or multiplications. These individuals are at risk of intoxication or exaggerated effects of tramadol due to higher concentrations of its active metabolite (M1). The occurrence of this phenotype is seen in approximately 1% to 2% of East Asians (Chinese, Japanese, Korean), 1% to 10% of Caucasians, 3% to 4% of African-Americans, and may be >10% in certain racial/ethnic groups (ie, Oceanian, Northern African, Middle Eastern, Ashkenazi Jews, Puerto Rican). The FDA label recommends avoiding the use of tramadol in these individuals.
Properties of Tramadol
| Melting point: | 178-181 °C |
| Boiling point: | 406.62°C (rough estimate) |
| Density | 0.9903 (rough estimate) |
| refractive index | 1.4909 (estimate) |
| storage temp. | 2-8°C |
| form | solid |
| pka | 14.47±0.40(Predicted) |
| color | white to off-white |
| CAS DataBase Reference | 27203-92-5(CAS DataBase Reference) |
| NIST Chemistry Reference | Tramadol(27203-92-5) |
| EPA Substance Registry System | Cyclohexanol, 2-[(dimethylamino)methyl]-1-(3-methoxyphenyl)-, (1R,2R)-rel- (27203-92-5) |
Safety information for Tramadol
Computed Descriptors for Tramadol
Tramadol manufacturer
New Products
4,4-Difluoropiperidine hydrochloride tert-butyl 9-methoxy-3-azaspiro[5.5]undecane-3-carboxylate Indole Methyl Resin N-Isopropylurea N,N-Dicyclohexylcarbodiimide(DCC) MELDRUMS ACID 5-METHYLISOXAZOLE-4-CARBOXYLIC ACID Magnessium Bis glycinate Zinc ascorbate 1-bromo-2-butyne 2-acetamidophenol 9(10H)-anthracenone Erythrosin B, 4-Piperidinopiperidine 2-((4-morpholinophenylamino) (methylthio) methylene) malononitrile 2,4-dihydroxybenzaldehyde 3-(4-morpholinophenylamino)-5-amino-1H-pyrazole-4-carbonitrile Methyl 2-methylquinoline-6-carboxylate 2,6-dichloro-4-nitropyridine 4-Bromo-2-chlorobenzonitrile 2-(benzylamino)acetic acid hydrochloride 4-(tert-Butoxycarbonylamino)but- 2-ynoic acid 3,4-dihydro-2H-benzo[b][1,4]dioxepine 1-Phenyl-1-cycloprppanecarboxylicacidRelated products of tetrahydrofuran


You may like
-
 3-(4-amino-1-oxoisoindolin-2-yl)-1-methylpiperidine-2,6-dione 98%View Details
3-(4-amino-1-oxoisoindolin-2-yl)-1-methylpiperidine-2,6-dione 98%View Details -
1-methylindoline-2,3-dione 98%View Details
2058-74-4 -
614-19-7 98%View Details
614-19-7 -
3112-85-4 Methyl phenyl sulfone 98%View Details
3112-85-4 -
20677-73-0 (2,2-diethoxyethyl)methylamine 98%View Details
20677-73-0 -
3-(4-(hydroxyamino)-1-oxoisoindolin-2-yl)piperidine-2,6-dione 98%View Details
-
57381-49-4 2-bromo-4-chlorobenzonitrile 98%View Details
57381-49-4 -
4,6-dichloropyrimidine-5-carbaldehyde 98%View Details
5305-40-8