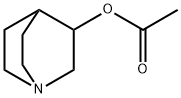
Aceclidine
- CAS NO.:827-61-2
- Empirical Formula: C9H15NO2
- Molecular Weight: 169.22
- MDL number: MFCD00468105
- EINECS: 212-574-1
- SAFETY DATA SHEET (SDS)
- Update Date: 2024-11-19 23:02:33
What is Aceclidine?
Originator
Glacostat ,MSD-Chibret ,France ,1966
The Uses of Aceclidine
analgesic (topical), depletes Substance P, neurotoxic
Background
Aceclidine has been marketed in Europe but has not been used clinically in the United States. It is used in the treatment of open-angle glaucoma and is a parasympathomimetic agent.
Definition
ChEBI: Acetic acid 1-azabicyclo[2.2.2]octan-3-yl ester is a member of quinuclidines.
Manufacturing Process
A mixture of 274 g of methyl isonicotinate, 367 g of ethyl bromoacetate and
125 cc of ethyl alcohol was stirred without heating for 4 hours in a flask
equipped with a reflux condenser. (The reaction was exothermic and
precautions were taken to keep the temperature below 70°C.) The reaction
mixture was then left for 15 hours at room temperature.
The reaction product (1-carbethoxymethyl-4-carbornethoxy-pyridinium
bromide) was obtained in crystalline form. (It formed prisms melting at 166-
169°C after recrystallization from a mixture of isopropanol and acetone.) It
was not necessary to isolate it. For the following reduction step, the reaction
mixture was brought into solution by the addition of about 1 liter of warm
ethyl alcohol. It was then hydrogenated at about 30 atm pressure in the
presence of 2 g of platinum oxide. The temperature rose during this reaction
to about 40°C. After the calculated amount of hydrogen had been absorbed,
the catalyst was filtered off, the solution was concentrated in vacuum, and the
residual syrup was dissolved in ice water. Benzene was added and the mixture
was made alkaline with an excess of concentrated ice cold potassium
carbonate solution. The temperature was kept low by continuous addition of
ice, and the benzene layer was separated and dried with sodium sulfate. The
dried benzene solution was concentrated in vacuum and the residual oil was
distilled in vacuum. BP 30 mm = 175-182°C, nD
25= 1.4613-1.4628. During
the reduction, partial alcoholysis occurred, and the product isolated was 1-
carbethoxymethyl-4-"carbalkoxy"-piperidine, wherein "carbalkoxy" represents
a mixture of carbomethoxy and carbethoxy.
100 g of potassium were pulverized in 200 cc of hot toluene in a heated
three-neck flask equipped with an efficient condenser, stirrer and dropping
funnel. To the refluxing potassium suspension were added in small portions
229 g of the product of the previous step and about 700 cc of toluene. This
addition had to be carried out very cautiously; the onset of the exothermic
reaction is sometimes delayed. The addition was finished in about 1 hour. To
complete the reaction, the refluxing and stirring were continued for about 4
hours. The reaction mixture was then cooled to about +5°C and about 50 cc
isopropanol were added to decompose unreacted potassium. Then 2.5 liters of
concentrated hydrochloric acid were added and the mixture was refluxed for
15 hours, and then concentrated in vacuum to dryness. To the residue was
added with cooling an excess of 50% potassium hydroxide. Ether was then
added and the resulting mixture was filtered through a fritted glass funnel,
thus removing the precipitated potassium chloride. The ethereal and aqueous
layers were separated, and the aqueous layer was extracted repeatedly with
500 cc portions of ether. The organic solutions were combined, dried over sodium sulfate and concentrated in vacuum. Aqueous hydrochloric acid was
added to the residue until the solution became acid. The mixture was then
diluted with distilled water to about 300 cc, heated with decolorizing charcoal,
filtered and concentrated in vacuum to dryness. The residue was treated with
isopropanol, and the precipitated crystalline product was filtered off. The
product was recrystallized from a mixture of water and isopropanol and was
identified as 1-azabicyclo[2.2.2]-3-octanone hydrochloride; prisms, MP 311-
313°C, with decomposition.
A solution of 50 g of the above ketone-hydrochloride in 30 cc of water was
made alkaline by the addition of 30 g of potassium hydroxide. After the alkali
was dissolved, 35 g of granular potassium carbonate were added. The free
basic ketone was then extracted from the viscous mixture by shaking with 4
portions of hot benzene (300 cc in each portion). The benzene extracts were
decanted, filtered over sodium sulfate in order to remove any suspended
alkali, and concentrated in vacuum. The residual lszabicyclo[2.2.2]-3-octanone
was purified by sublimation (50-70°C/0.5 mm Hg); it can also be purified by
recrystallization from petroleum ether. It formed feathery crystals melting at
147-148°C.
The product was reduced as follows:
A solution of 50 g of 1-azabicyclo[2.2.2]-3-octanone hydrochloride in 200 cc
of water was hydrogenated at room temperature and 50 atm pressure with 1
g of platinum oxide as catalyst. After the calculated amount of hydrogen had
been absorbed, the mixture was filtered and concentrated in vacuum to
dryness. The residual product was recrystallized from a mixture of methanol
and acetone and formed prisms melting above 300°C. It was identified as 1-
azabicyclo[2.2.2]-3-octanol hydrochloride.
A solution of 50 g of 1-azabicyclo[2.2.2]-3-octanol hydrochloride in 30 cc
water was made alkaline with 30 g of potassium hydroxide. After the alkali
was dissolved 35 g of granular potassium carbonate were added. The free
basic alcohol was then extracted from the viscous mixture by shaking with
four portions of boiling benzene (300 cc in each portion). The benzene
extracts were decanted and filtered over anhydrous sodium sulfate, to remove
any suspended alkali. The combined benzene solutions were concentrated in
vacuum. The residue was recrystallized from benzene and identified as
lszabicyclo[2.2.2]-3-octanol, MP 221-223°C. The product can also be purified
by recrystallization from acetone, or by sublimation in vacuum (120°C/20 mm
Hg). The alcohol was reacted with acetic anhydride to give the product
aceclidine.
brand name
Cholinergic Glaucostat (Kingshill Pharmaceuticals, Inc, Switzerland).
Therapeutic Function
Miotic, Cholinomimetic
Safety Profile
Poison by ingestion, subcutaneous,and intravenous routes. When heated to decomposition itemits toxic fumes of NOx.
Metabolism
Not Available
Properties of Aceclidine
| Boiling point: | 298.52°C (rough estimate) |
| Density | 1.0873 (rough estimate) |
| refractive index | 1.4780 (estimate) |
| storage temp. | Sealed in dry,Store in freezer, under -20°C |
| pka | 9.22±0.33(Predicted) |
| form | <34°C Solid,>36°C Liquid |
| color | Off-white to light yellow |
Safety information for Aceclidine
Computed Descriptors for Aceclidine
New Products
Tert-butyl bis(2-chloroethyl)carbamate 4-Methylphenylacetic acid N-Boc-D-alaninol N-BOC-D/L-ALANINOL 3-Morpholino-1-(4-nitrophenyl)-5,6-dihydropyridin- 2(1H)-one Furan-2,5-Dicarboxylic Acid Tropic acid 1,1’-CARBONYLDIIMIDAZOLE DIETHYL AMINOMALONATE HYDROCHLORIDE R-2-BENZYLOXY PROPIONIC ACID 1,1’-CARBONYLDI (1,2-4 TRIAZOLE) N-METHYL INDAZOLE-3-CARBOXYLIC ACID (2-Hydroxyphenyl)acetonitrile 4-Bromopyrazole 5-BROMO-2CYANO PYRIDINE 5,6-Dimethoxyindanone 5-broMo-2-chloro-N-cyclopentylpyriMidin-4-aMine 2-(Cyanocyclohexyl)acetic acid 4-methoxy-3,5-dinitropyridine 2-aminopropyl benzoate hydrochloride 1-(4-(aminomethyl)benzyl)urea hydrochloride diethyl 2-(2-((tertbutoxycarbonyl)amino) ethyl)malonate tert-butyl 4- (ureidomethyl)benzylcarbamate Ethyl-2-chloro((4-methoxyphenyl)hydrazono)acetateRelated products of tetrahydrofuran


You may like
-
 Quinuclidin-3-yl acetate 95% CAS 827-61-2View Details
Quinuclidin-3-yl acetate 95% CAS 827-61-2View Details
827-61-2 -
1975-50-4 98%View Details
1975-50-4 -
2-HYDROXY BENZYL ALCOHOL 98%View Details
90-01-7 -
2-Chloro-1,3-Bis(Dimethylamino)Trimethinium Hexafluorophosphate 221615-75-4 98%View Details
221615-75-4 -
61397-56-6 CIS BROMO BENZOATE 98%View Details
61397-56-6 -
14714-50-2 (2-Hydroxyphenyl)acetonitrile 98+View Details
14714-50-2 -
118753-70-1 98+View Details
118753-70-1 -
733039-20-8 5-broMo-2-chloro-N-cyclopentylpyriMidin-4-aMine 98+View Details
733039-20-8