
10-Propargyl-10-deazaaminopterin
- CAS NO.:146464-95-1
- Empirical Formula: C23H23N7O5
- Molecular Weight: 477.47
- MDL number: MFCD00920897
- SAFETY DATA SHEET (SDS)
- Update Date: 2025-12-23 21:30:31
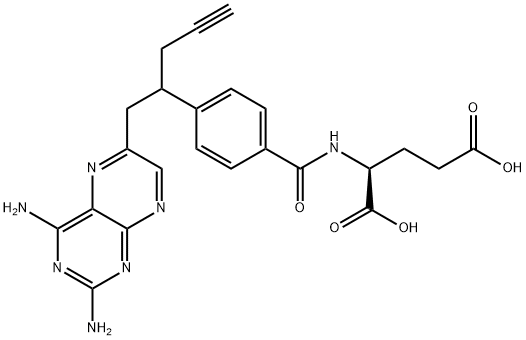
What is 10-Propargyl-10-deazaaminopterin?
Absorption
With an intravenous formulation, pralatrexate has complete bioavailability. Pralatrexate demonstrates a dose-proportional and linear pharmacokinetics over a dose range of 30-325 mg/m2. Upon an intravenous push over 3 to 5 minutes of a starting dose of 30 mg/m2 racemic pralatrexate for dose 1 of cycle 1, Cmax and AUC0-∞ was estimated to be 5,815 ng/mL and 267,854 ng/mL.min respectively using a noncomparmental pharmacokinetics analysis.Both pralatrexate diastereomers demonstrates a multiphase decline in plasma concentration with a rapid initial fall followed by a slow terminal phase. The initial fall is thought to reflect the clearance of pralatrexate by renal and non-renal mechanism , while the slow terminal phase likely represents the return of pralatrexate from deep intracellular compartments, enterohepatic circulation, or after deglutamination.
Toxicity
Mucositis is dose-limiting toxicity. Folic acid and vitamin B12 supplements do not prevent mucositis from happening.
No specific information is available on the treatment of overdosage of pralatrexate. If an overdose occurs, general supportive measures should be instituted as deemed necessary by the treating healthcare provider. Based on pralatrexate's mechanism of action, consider the prompt administration of leucovorin.
Carcinogenicity studies and fertility studies have not been performed with pralatrexate.Based on findings from animal studies and its mechanism of action [see Clinical Pharmacology (12.1)], pralatrexate can cause fetal harm when administered to a pregnant woman. There are insufficient data on pralatrexate use in pregnant women to evaluate for a drug-associated risk. Pralatrexate was embryotoxic and fetotoxic in rats and rabbits when administered during organogenesis at doses about 1.2% (0.012 times) of
the clinical dose on an mg/m2 basis. Advise pregnant women of the potential risk to a fetus.
Pralatrexate did not cause mutations in the Ames test or the Chinese hamster ovary cell chromosome aberration assay. Nevertheless, these tests do not reliably predict genotoxicity for this class of compounds. Pralatrexate did not cause mutations in the mouse micronucleus assay.
Description
Pralatrexate, an injectable DHFR inhibitor, was launched for the treatment of patients with relapsed or refractory PTCL. PTCL is an aggressive form of non-Hodgkin’s lymphoma (NHL) characterized by the proliferation of abnormal T-lymphocytes that circulate in the peripheral bloodstream. The inhibition of the folate enzymes DHFR and thymidylate synthase is a well-validated method of cancer treatment. In vitro, pralatrexate is slightly less potent than MTX in inhibiting DHFR derived from murine leukemia L1210 cells (Ki = 18.2 pM vs. 5.75 pM) and human leukemia CCRF-CEM cells (Ki = 13.4 pM vs. 5.4 pM). However, it is transported into both types of cells with 10-fold higher efficiency than MTX, thereby providing a more potent inhibition of cell growth as compared with MTX. In vivo, intraperitonally administered pralatrexate at 60 mg/ kg twice weekly for three or four doses caused complete lymphoma regressions in 89, 56, and 30% of HT, RL, and SKI-DLBCL-1 xenografted mice, respectively, whereas a similar dosing of MTX at 40 mg/kg twice weekly did not produce complete regression. The posttreatment tumor diameter was also smaller in pralatrexate-treated animals.
Description
Pralatrexate is a dihydrofolate reductase (DHFR) inhibitor (Ki = 13.4 pM) and antifolate. It inhibits growth of CCRF-CEM acute lymphocytic leukemia cells (IC50 = 0.04 μM), MDA-468, SK-BR-3, and ZR-75-1 breast cancer cells (IC50s = 0.11, 0.28, and 0.26 μM, respectively), and SK-LC8 and SK-LC16 non-small cell lung cancer cells (NSCLC; IC50s = 0.42 and 0.11 μM, respectively). In vivo, pralatrexate increases median survival from 21 to 40 days when administered in 4 doses of 15 mg/kg over 11 days in an H9 T cell lymphoma mouse xenograft model. Pralatrexate is transported into cells via the reduced folate carrier (RFC) and undergoes polyglutamation by folylpolyglutamate synthetase (FPGS) to a greater extent than methotrexate or pemetrexed . Formulations containing pralatrexate have been used in the treatment of relapsed or refractory peripheral T cell lymphoma.
Originator
SRI International/ Southern Research Institute/Sloan-Kettering (US)
The Uses of 10-Propargyl-10-deazaaminopterin
An antifolate with high affinity for the reduced folate carrier-type 1, produces marked complete and durable remissions in a diversity of chemotherapy refractory cases of T-cell lymphoma.
Background
Pralatrexate is an antifolate for the treatment of relapsed or refractory peripheral T-cell lymphoma . Pralatrexate was developed in response due to the inferior responses of patients using the standard therapy for their B-cells counterparts. Compared to methotrexate, pralatrexate has better accumulation in cancer cells. Pralatrexate is designed to have a higher affinity for the reduced folate carrier, a protein that is overexpressed in malignant cells and is upregulated by oncogenes. As such, pralatrexate is thought to have a better therapeutic window compared to other antifolate analogs due to the novel target of RFC.
Pralatrexate was approved by the FDA on September 24, 2009. It is also being studied for other types of lymphoma and solid malignancy such as non-small-cell lung cancer, breast cancer, and bladder cancer.
Indications
Pralatrexate is indicated for the treatment of relapsed or refractory peripheral T-cell lymphoma.
What are the applications of Application
Folotyn is an inhibitor of DHFR (dihydrofolate reductase)
Definition
ChEBI: A pteridine that is the N-4-[1-(2,4-diaminopteridin-6-yl)pent-4-yn-2-yl]benzoyl derivative of L-glutamic acid. Used for treatment of Peripheral T-Cell Lymphoma, an aggressive form of non-Hodgkins lymphoma.
brand name
Folotyn
Pharmacokinetics
Pralatrexate is a folate analog that inhibits folate metabolism, thus impeding the synthesis of amino acids and nucleic acid. Additionally, pralatrexate also competes for enzymatic processing by folyopolyglutamate synthase (FPGS)with folate to increase cellular retention. Compared to methotrexate, pralatrexate binds to the reduced folate carrier protein-1 (RFC-1) for cellular uptake with 10-times the affinity and is a more potent substrate for FPGS. The Km value for RFC-1 was calculated to be 0.3 μmol/L and 4.8 μmol/L for pralatrexate and methotrexate respectively, while the Km value for FPGS was estimated to be 5.9 and 32.3 μmol/l for pralatrexate and methotrexate respectively. As a result, pralatrexate is more cytotoxic and better retained in cancer cells.
Due to its anti-folate activity, pralatrexate's main toxicity is manifested as mucositis that can require dose interruption or reduction. In 5 patients with non-small-cell lung carcinoma receiving a supratherapeutic dose of 230 mg/m2, the mean change from pre-injection QTcF interval at the end of infusion was 6.1 ms (90%CI: -0.6, 12.7), and at 1-hour post-injection was 7.8 ms (90%CI: 3.0, 12.6). However, no patient exceeded a QTcF of 470 msec and exhibited an absolute increase from baseline in QTcF exceeding 30 msec. As well, the study dose far exceeded the target dose for patients with peripheral T-cell lymphoma and pralatrexate does not inhibit the human ether-a-go-go-related gene (hERG) K+ channel. Therefore, pralatrexate uses are unlikely to cause cardiac repolarization delays..
Clinical Use
Pralatrexate, an injectable dihydrofolate reductase (DHFR) inhibitor, has a superior potency and toxicity profile compared to other DHFR inhibitors. In 2009, the compound was launched by Allos and approved in the U.S. for the treatment of patients with relapsed or refractory peripheral T-cell lymphoma (PTCL) as a single agent. It is the first drug approved for this indication.70 In 2010, orphan drug designation was received in the E.U. for the treatment of cutaneous T-cell lymphoma (CTCL).
Side Effects
The most common adverse reactions associated with pralatrexate are mucositis, thrombocytopenia, nausea, and fatigue. Folic acid and vitamin B12 supplements are administered as adjunct therapies to potentially reduce pralatrexate-related hematological toxicity and mucositis.
Synthesis
The chemical synthesis of pralatrexate starts with the alkylation of the anion of dimethyl homoterephthalate with propargyl bromide, promoted by potassium hydride in dimethylformamide, to afford the corresponding a-propargyl diester. Further alkylation of the potassium salt of a-propargyl diester with 2,4-diamino-6-(bromomethyl)pteridine followed by saponification with sodium hydroxide yields a diacid intermediate (2,4diamino- 4-deoxy-10-propargyl-10-deazapteroic acid). Mono-decarboxylation of the diacid intermediate by heating in dimethylsulfoxide at 120 C, followed by coupling with diethyl L-glutamate, and subsequent ester hydrolysis with sodium hydroxide yields pralatrexate.
Metabolism
While the liver has been shown to metabolize pralatrexate to some extent, pralatrexate is not significantly metabolized by any CYP450 isozymes or glucuronidases in vitro.
References
[1]. izbicka e, diaz a, streeper r, et al. distinct mechanistic activity profile of pralatrexate in comparison to other antifolates in in vitro and in vivo models of human cancers. cancer chemother pharmacol, 2009, 64(5): 993-999.
[2]. serova m, bieche i, sablin mp, et al. single agent and combination studies of pralatrexate and molecular correlates of sensitivity. br j cancer, 2011, 104(2): 272-280.
Properties of 10-Propargyl-10-deazaaminopterin
| Melting point: | 215 °C(dec.) |
| Density | 1.471±0.06 g/cm3(Predicted) |
| storage temp. | 2-8°C |
| Water Solubility | Insoluble in water |
| solubility | ≥23.85 mg/mL in DMSO; insoluble in H2O; insoluble in EtOH |
| pka | 3.53±0.10(Predicted) |
| form | powder to crystal |
| color | White to Light yellow |
Safety information for 10-Propargyl-10-deazaaminopterin
| Signal word | Warning |
| Pictogram(s) |
 Exclamation Mark Irritant GHS07  Health Hazard GHS08 |
| GHS Hazard Statements |
H302:Acute toxicity,oral H361:Reproductive toxicity |
| Precautionary Statement Codes |
P201:Obtain special instructions before use. P202:Do not handle until all safety precautions have been read and understood. P264:Wash hands thoroughly after handling. P264:Wash skin thouroughly after handling. P270:Do not eat, drink or smoke when using this product. P280:Wear protective gloves/protective clothing/eye protection/face protection. P308+P313:IF exposed or concerned: Get medical advice/attention. P405:Store locked up. P501:Dispose of contents/container to..… |
Computed Descriptors for 10-Propargyl-10-deazaaminopterin
10-Propargyl-10-deazaaminopterin manufacturer
ALS India Life Sciences Pvt. Ltd
New Products
4,4-Difluoropiperidine hydrochloride tert-butyl 9-methoxy-3-azaspiro[5.5]undecane-3-carboxylate Indole Methyl Resin N-Isopropylurea N,N-Dicyclohexylcarbodiimide(DCC) MELDRUMS ACID 5-METHYLISOXAZOLE-4-CARBOXYLIC ACID Magnessium Bis glycinate Zinc ascorbate 1-bromo-2-butyne 2-acetamidophenol 9(10H)-anthracenone Erythrosin B, 4-Piperidinopiperidine 2-((4-morpholinophenylamino) (methylthio) methylene) malononitrile 2,4-dihydroxybenzaldehyde 3-(4-morpholinophenylamino)-5-amino-1H-pyrazole-4-carbonitrile Methyl 2-methylquinoline-6-carboxylate 2,6-dichloro-4-nitropyridine 4-Bromo-2-chlorobenzonitrile 2-(benzylamino)acetic acid hydrochloride 4-(tert-Butoxycarbonylamino)but- 2-ynoic acid 3,4-dihydro-2H-benzo[b][1,4]dioxepine 1-Phenyl-1-cycloprppanecarboxylicacidRelated products of tetrahydrofuran
You may like
-
 146464-95-1 Pralatrexate 98%View Details
146464-95-1 Pralatrexate 98%View Details
146464-95-1 -
 146464-95-1 98%View Details
146464-95-1 98%View Details
146464-95-1 -
 Pralatrexate CAS 146464-95-1View Details
Pralatrexate CAS 146464-95-1View Details
146464-95-1 -
146464-95-1 95.00%View Details
146464-95-1 -
3-(4-amino-1-oxoisoindolin-2-yl)-1-methylpiperidine-2,6-dione 98%View Details
-
20677-73-0 (2,2-diethoxyethyl)methylamine 98%View Details
20677-73-0 -
3-(4-(hydroxyamino)-1-oxoisoindolin-2-yl)piperidine-2,6-dione 98%View Details
-
57381-49-4 2-bromo-4-chlorobenzonitrile 98%View Details
57381-49-4