
XL518
- CAS NO.:934660-93-2
- Empirical Formula: C21H21F3IN3O2
- Molecular Weight: 531.31
- MDL number: MFCD22124461
- SAFETY DATA SHEET (SDS)
- Update Date: 2025-12-26 16:58:18
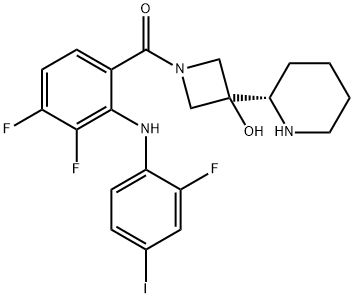
What is XL518?
Absorption
Following oral dosing of 60 mg once daily in cancer patients, the median time to achieve peak plasma levels (Tmax) was 2.4 (range:1–24) hours, geometric mean steady-state AUC0-24h was 4340 ng?h/mL (61% CV) and Cmax was 273 ng/mL (60% CV). The absolute bioavailability of COTELLIC was 46% (90% CI: 40%, 53%) in healthy subjects. A high‐fat meal (comprised of approximately 150 calories from protein, 250 calories from carbohydrate, and 500–600 calories from fat) had no effect on cobimetinib AUC and Cmax after a single 20 mg COTELLIC was administered to healthy subjects.
Toxicity
The most common adverse effects (>20%) for cobimetinib are diarrhea, photosensitivity reactions, nausea, fever and vomiting.
Description
Cobimetinib, codeveloped by Genentech and Exelixis, was approved in August 2015 in Switzerland and November 2015 in the U.S. and Europe for the treatment of unresectable or metastatic BRAFV600 mutationpositive melanoma when used in combination with vemurafenib. Cobimetinib is a potent, highly selective reversible inhibitor of mitogen-activated protein kinases (MEK) 1 and 2,120 which serves to inhibit phosphorylation of ERK1/2,121 disrupting the MAPK pathway which is responsible for cell proliferation, cell survival, and migration.122 Combination of cobimetinib with vemurafenib, an important BRAF inhibitor,123 enables targeting of multiple points on the MAPK pathway, leading to overall enhanced tumor cell apoptosis and response as compared to stand-alone treatment with vemurafenib.124 Specifically, in a representative trial of previously untreated patients with BRAFV600 mutation-positive, unresectable, stage IIIc or IV melanoma, combination of these two therapies led to a significantly improved progression-free survival and overall response rate versus patients treated only with vemurafenib.
The Uses of XL518
A potent, selective, orally bioavailable inhibitor of MEK1, a component of the RAS/RAF/MEK/ERK pathway. It inhibits proliferation and stimulates apoptosis in a variety of human tumor cell lines. In preclinical xenograft models, oral administration of XL518 results in sustained inhibition of pERK in tumor tissue, but not brain tissue, leading to tumor growth inhibition and regression at well tolerated doses.
Background
Cobimetinib is an orally active, potent and highly selective small molecule inhibiting mitogen-activated protein kinase kinase 1 (MAP2K1 or MEK1), and central components of the RAS/RAF/MEK/ERK signal transduction pathway. It has been approved in Switzerland and the US, in combination with vemurafenib for the treatment of patients with unresectable or metastatic BRAF V600 mutation-positive melanoma.
Indications
Cobimetinib is indicated in combination with vemurafenib for the treatment of unresectable or metastatic melanoma with a BRAF V600E or V600K mutation. As a single agent, cobimetinib is also indicated for the treatment of adult patients with histiocytic neoplasms.
Definition
ChEBI: Cobimetinib is a member of the class of N-acylazetidines obtained by selective formal condensation of the carboxy group of 3,4-difluoro-2-(2-fluoro-4-iodoanilino)benzoic acid with the secondary amino group from the azetidine ring of 3-[(2S)-piperidin-2-yl]azetidin-3-ol. An inhibitor of mitogen-activated protein kinase that is used (as its fumarate salt) in combination with vemurafenib for the treatment of patients with unresectable or metastatic melanoma. It has a role as an EC 2.7.11.24 (mitogen-activated protein kinase) inhibitor and an antineoplastic agent. It is a member of piperidines, a N-acylazetidine, a tertiary alcohol, an aromatic amine, a secondary amino compound, a difluorobenzene and an organoiodine compound. It is a conjugate base of a cobimetinib(1+).
brand name
Cotellic
General Description
Class: dual threonine/tyrosine kinase; Treatment: melanoma with BRAF mutations; Other name: GDC-0973, XL518; Oral bioavailability = 46%; Elimination half-life = 44 h; Protein binding = 95%
Pharmacokinetics
Cobimetinib is a reversible inhibitor of mitogen-activated protein kinase 1 (MAPK)/extracellular signal-regulated kinase 1 (MEK1) and MEK2. Preclinical studies have demonstrated that this agent is effective in inhibiting the growth of tumor cells bearing a BRAF mutation, which has been found to be associated with many tumor types. A threonine-tyrosine kinase and a key component of the RAS/RAF/MEK/ERK signaling pathway that is frequently activated in human tumors, MEK1 is required for the transmission of growth-promoting signals from numerous receptor tyrosine kinases. Cobimetinib is used in combination with vemurafenib because the clinical benefit of a BRAF inhibitor is limited by intrinsic and acquired resistance. Reactivation of the MAPK pathway is a major contributor to treatment failure in BRAF-mutant melanomas, approximately ~80% of melanoma tumors become BRAF-inhibitor resistant due to reactivation of MAPK signaling. BRAF-inhibitor-resistant tumor cells are sensitive to MEK inhibition, therefore cobimetinib and vemurafenib will result in dual inhibition of BRAF and its downstream target, MEK.
Pharmacokinetics
Cobimetinib has only moderate oral
bioavailability (46%), likely due to metabolism rather
than incomplete absorption. However, it displays
prolonged elimination half-life (44 h), which supports
a once-daily dosing regimen (60 mg). Following oral administration, the unchanged
cobimetinib and metabolite 4 were the major
circulating components in the plasma up to 48 hours
post dose (AUC0–48), accounting for 21% and 18% of
all the circulating drug-related components,
respectively (Fig. 4).
Clinical Use
Protein kinase inhibitor:
Treatment of unresectable or metastatic melanoma
with a BRAF V600 mutation in combination with
vemurafenib
Synthesis
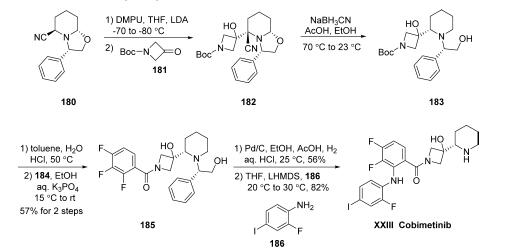
Structurally, cobimetinib features an interesting azetidinol substructure appended to the 2-position of a piperidine, rendering the 2-carbon of the piperidine as a stereogenic center bearing the (S)-configuration. While the early discovery routes to cobimetinib relied on a piperidine resolution-based route for accessing the cobimetinib core, the scale route to this drug employs an impressive N-cyanomethyl oxazolidine chiral auxiliary-mediated sequence to induce strereocontrol, generating the requisite stereocenter with excellent selectivity and requiring no chromatographic purification in the overall synthetic sequence. Toward this end, the most likely scale synthetic approach was initiated with deprotonation of commercially available (3S,5R,8aS)-3-phenyl-hexahydrooxazolo[ 3,2-a]pyridine-carbonitrile (180), followed by addition of commercial 3-oxo-azetidine-1-carboxylic acid tert-butyl ester (181), yielded 182 in high purity (92%) after distillation and providing a rapid route to the core structure of cobimetinib. One-pot ring opening and reduction of 182 was accomplished by exposing this hemiaminal to acetic acid and sodium cyanoborohydride, giving rise to intermediate 183. This carbamate could be further reacted with aqueous HCl in toluene to liberate the azetidine amine salt in high purity (97.6%), which underwent immediate acylation with commercially available 2,3,4-trifluoro-benzoyl chloride (184) to enable formation of intermediate 185 in 85% purity after aqueous workup. Reductive cleavage of the chiral auxiliary of 185 with Pd/C and H2 under aqueous acidic conditions (AcOH, aq HCl) yielded the desired piperidine amine, which could be isolated as a solid (99.6% pure) after trituration with aqueous HCl. Finally, aromatic fluoride substitution with commercially available aniline 186 under basic conditions provided cobimetinib in 99.7% purity after slow precipitation from toluene (it is important to note that the authors offer no comment as to the regioselectivity of this aromatic substitution reaction). While the drug reportedly exists as a fumarate salt, no synthetic reports describing the conversion of cobimetinib to the corresponding fumarate salt were available in the chemical literature to our knowledge at the time of publication.
Drug interactions
Potentially hazardous interactions with other drugs
Antifungals: concentration increased by itraconazole.
Antipsychotics: increased risk of agranulocytosis -
avoid.
Metabolism
Cobimetinib is mainly metabolized via CYP3A oxidation and UGT2B7 glucuronidation with no major metabolites formed.
Metabolism
Metabolised by oxidation by CYP3A and glucuronidation by UGT2B7. Extensively metabolised and eliminated in faeces
Properties of XL518
| Melting point: | 165 - 166°C |
| Boiling point: | 565.9±50.0 °C(Predicted) |
| Density | 1.706 |
| storage temp. | Refrigerator |
| solubility | Chloroform (Slightly), DMSO (Slightly), Ethyl Acetate (Slightly), Methanol (Slig |
| form | Solid |
| pka | 13.13±0.20(Predicted) |
| color | Off-White |
Safety information for XL518
Computed Descriptors for XL518
New Products
4,4-Difluoropiperidine hydrochloride tert-butyl 9-methoxy-3-azaspiro[5.5]undecane-3-carboxylate Indole Methyl Resin N-Isopropylurea N,N-Dicyclohexylcarbodiimide(DCC) MELDRUMS ACID 5-METHYLISOXAZOLE-4-CARBOXYLIC ACID Magnessium Bis glycinate Zinc ascorbate 1-bromo-2-butyne 2-acetamidophenol 9(10H)-anthracenone Erythrosin B, 4-Piperidinopiperidine 2-((4-morpholinophenylamino) (methylthio) methylene) malononitrile 2,4-dihydroxybenzaldehyde 3-(4-morpholinophenylamino)-5-amino-1H-pyrazole-4-carbonitrile Methyl 2-methylquinoline-6-carboxylate 2,6-dichloro-4-nitropyridine 4-Bromo-2-chlorobenzonitrile 2-(benzylamino)acetic acid hydrochloride 4-(tert-Butoxycarbonylamino)but- 2-ynoic acid 3,4-dihydro-2H-benzo[b][1,4]dioxepine 1-Phenyl-1-cycloprppanecarboxylicacidRelated products of tetrahydrofuran

You may like
-
 3-(4-amino-1-oxoisoindolin-2-yl)-1-methylpiperidine-2,6-dione 98%View Details
3-(4-amino-1-oxoisoindolin-2-yl)-1-methylpiperidine-2,6-dione 98%View Details -
1-methylindoline-2,3-dione 98%View Details
2058-74-4 -
614-19-7 98%View Details
614-19-7 -
3112-85-4 Methyl phenyl sulfone 98%View Details
3112-85-4 -
20677-73-0 (2,2-diethoxyethyl)methylamine 98%View Details
20677-73-0 -
3-(4-(hydroxyamino)-1-oxoisoindolin-2-yl)piperidine-2,6-dione 98%View Details
-
57381-49-4 2-bromo-4-chlorobenzonitrile 98%View Details
57381-49-4 -
4,6-dichloropyrimidine-5-carbaldehyde 98%View Details
5305-40-8