
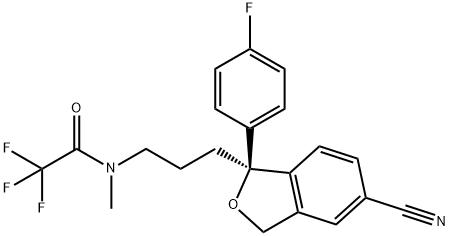
Escitalopram
- CAS NO.:128196-01-0
- Empirical Formula: C20H21FN2O
- Molecular Weight: 324.39
- MDL number: MFCD09817397
- SAFETY DATA SHEET (SDS)
- Update Date: 2025-12-17 11:34:44
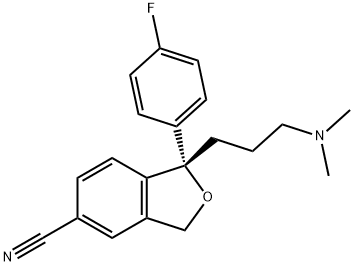
What is Escitalopram?
Absorption
Absorption of escitalopram following oral administration is expected to be almost complete, with an estimated absolute bioavailability of approximately 80%. Tmax occurs after about 4-5 hours. Cmax and AUC appear to follow dose proportionality - at steady state, patients receiving 10mg of escitalopram daily had a Cmax of 21 ng/mL and a 24h AUC of approximately 360 ng*h/mL, while patients receiving 30mg daily had a roughly 3-fold increase in both Cmax and 24h AUC, comparatively.
Toxicity
Symptoms of overdose may include CNS effects (dizziness, convulsions, coma, somnolence), gastrointestinal distress (nausea, vomiting), and/or cardiac abnormalities (hypotension, tachycardia, ECG changes). There is no specific antidote for escitalopram overdose. Management of overdose should focus on monitoring for cardiac abnormalities and changes to vital signs as well as treatment with supportive measures as indicated. As escitalopram is highly distributed into tissue following oral administration, forced diuresis, dialysis, and other methods of extracting drug from plasma are unlikely to be beneficial.
Description
Escitalopram is a selective serotonin uptake inhibitor marketed by Forest Laboratories in the United States as an antidepressant under the trade name Lexapro. Elsewhere it is sold by the Danish company Lundbeck under the names Ciraplex, Sipralexa, and Seroplex. It was codeveloped by the two companies. Escitalopram is the?S-isomer of the earlier Lundbeck drug citalopram (Celexa), and studies have established its superior performance over the enantiomeric mixture.
The Uses of Escitalopram
antidepressive;selective serotonin reuptake inhibitor
The Uses of Escitalopram
(S)-Citalopram is an inhibitor of serotonin (5-HT) uptake. Antidepressant.
Background
Escitalopram is a selective serotonin re-uptake inhibitor (SSRI) and the S-enantiomer of racemic citalopram. It is used to restore serotonergic function in the treatment of depression and anxiety. Escitalopram is approximately 150 times more potent than citalopram’s R-enantiomer and is responsible for the vast majority of citalopram’s clinical activity, with some evidence suggesting that the R-enantiomer of racemic citalopram actively dampens the activity of escitalopram rather than existing simply as an inactive enantiomer. Amongst SSRIs, escitalopram exerts the highest degree of selectivity for the serotonin transporter (SERT) relative to other off-targets which may explain its lower rates of adverse effects as compared to other agents in this class. Escitalopram also differentiates itself from other SSRIs via allosteric action on its target - this may be the mechanism responsible for its observed superior efficacy and faster onset compared to other SSRIs.
What are the applications of Application
Escitalopram is a selective serotonin reuptake inhibitor
Indications
Escitalopram is indicated for the acute and maintenance treatment of major depressive disorder (MDD) in adults and pediatric patients 12 years old and older and for the acute treatment of generalized anxiety disorder (GAD) in adults and pediatric patients 7 years old and older. It is additionally indicated for symptomatic relief of obsessive-compulsive disorder (OCD) in Canada.
Definition
ChEBI: A 1-[3-(dimethylamino)propyl]-1-(4-fluorophenyl)-1,3-dihydro-2-benzofuran-5-carbonitrile that has S-configuration at the chiral centre. It is the active enantiomer of citalopram.
Biological Activity
escitalopram (lexapro,cipralex)is a selective inihibitor of serotonin(5-ht) reuptake (ssri) with a ki value of 6.6nm for [3h]-5-ht uptake [1].escitalopram is the s-(+)-enantiomer of citalopram and has the inhibition of [3h]-5-ht uptake and [125i]-rti-55 binding in cos-1 cells expressing the human serotonin transporter (5-htt) with ki values of 6.6±1.4nm and 3.9±2.2nm, respectively. in addition, escitalopram has been reported to inhibit the accumulation of 3h-labelled monoamines into rat brain synaptosomes in vitro with ic50 values of 2.1±0.75nm, 2500±202nm and 40000±15500nm for [3h]-5-ht, [3h]-i-na and [3h]-da, respectively. apart from these, escitalopram has shown the in vitro affinity for rat histamine h1 receptors and the rat sigma σ1 site with ki values of 1500±780nm and 100±71nm, respectively [1].
Mechanism of action
Escitalopram is the S-enantiomer of citalopram that binds with high affinity and selectivity to the human SERT equivalent to (±)-citalopram. It has been reported that nearly all the activity resides in the S-enantiomer and that R-citalopram actually counteracts the action of the S-enantiomer. Studies show that escitalopram exhibits twice the activity of citalopram and is at least 27 times more potent than the R-enantiomer. The R-enantiomer inhibits the S-enantiomer at the transporter. Escitalopram's mechanism of action is common to the SSRIs.
Pharmacokinetics
Escitalopram belongs to a class of medications called selective serotonin re-uptake inhibitors (SSRIs). These agents cause an increase in serotonin levels in neuronal synapses by preventing the re-uptake of serotonin (5-HT) into the presynaptic terminals of serotonergic neurons. As compared to other SSRIs, it appears to have a relatively quick onset of effect due to its potency.
SSRIs as a class have been associated with abnormal bleeding, particularly in patients receiving concomitant therapy with other medications affecting hemostasis, and with the development of serotonin syndrome. Use escitalopram with caution in patients with a higher-than-baseline risk of bleeding and in patients receiving concomitant therapy with other serotonergic drugs. Escitalopram may also cause a discontinuation syndrome with abrupt removal of the drug, and should be slowly tapered if discontinuation of therapy is warranted.
Pharmacokinetics
The pharmacokinetics for escitalopram does not exhibit stereoisomer selectivity and, therefore, is similar to that for citalopram. Likewise, it exhibits linear pharmacokinetics so that plasma levels increase proportionately and predictably with increased doses, and its half-life of 27 to 32 hours is consistent with once-daily dosing. It also has been found that R-citalopram is cleared more slowly than the S-enantiomer. Therefore, when the drug is used as a racemic mixture (citalopram), the inactive isomer predominates at steady state. This is an added incentive for use of the enantiomerically pure escitalopram. Escitalopram has negligible effects on CYP isoforms, suggesting a low potential for drug–drug interactions. Escitalopram is indicated for patients with major depressive disorder, generalized anxiety disorder, panic disorder, and social anxiety disorder.
Clinical Use
SSRI antidepressant:
Depressive illness
Panic and social anxiety disorder
Drug interactions
Potentially hazardous interactions with other drugsAnalgesics: increased risk of bleeding with aspirin and NSAIDs; risk of CNS toxicity increased with tramadol.Anti-arrhythmics: increased risk of ventricular arrhythmias with amiodarone, disopyramide and dronedarone - avoid.Antibacterials: increased risk of ventricular arrhythmias with IV erythromycin, moxifloxacin, pentamidine and telithromycin.Anticoagulants: effect of coumarins possibly enhanced; possibly increased risk of bleeding with dabigatran.Antidepressants: avoid concomitant use with MAOI, increased risk of toxicity; increased risk of CNS toxicity with moclobemide - avoid concomitant use; avoid concomitant use with St John’s wort; possibly enhanced serotonergic effects with dapoxetine and duloxetine; can increase concentration of tricyclics; increased agitation and nausea with tryptophan; increased risk of CNS toxicity with rasagiline; possible increased risk of convulsions with vortioxetineAntiepileptics: convulsive threshold lowered.Antihistamines: increased risk of ventricular arrhythmias with mizolastine - avoid.Antimalarials: avoid concomitant use with artemether/lumefantrine and piperaquine with artenimol; possible increased risk of ventricular arrhythmias with chloroquine and quinine.Antipsychotics: possibly increased risk of ventricular arrhythmias with haloperidol, phenothiazines and pimozide - avoid.Antivirals: concentration possibly increased by ritonavir.Beta-blockers: increased risk of ventricular arrhythmias with sotalol - avoid.Dopaminergics: avoid with selegiline; increased risk of CNS toxicity with rasagiline.5HT1 agonist: increased risk of CNS toxicity with sumatriptan; possibly increased risk of serotonergic effects with naratriptan.Linezolid: use with care, possibly increased risk of side effects.Lithium: increased risk of CNS effectsMethylthioninium: risk of CNS toxicity - avoid if possible.
Metabolism
The metabolism of escitalopram is mainly hepatic, mediated primarily by CYP2C19 and CYP3A4 and, to a lesser extent, CYP2D6. Oxidative N-demethylation by the CYP enzyme system results in S-desmethylcitalopram (S-DCT) and S-didesmethylcitalopram (S-DDCT) - these metabolites do not contribute to the pharmacologic activity of escitalopram, and exist in the plasma in small quantities relative to the parent compound (28-31% and <5%, respectively).
There is also some evidence that escitalopram is metabolized to a propionic acid metabolite by monoamine oxidase A and B in the brain, and that these enzymes constitute the major route of escitalopram metabolism in the brain.
Metabolism
Escitalopram is metabolised in the liver to the demethylated and didemethylated metabolites. Both of these are pharmacologically active. Alternatively, the nitrogen may be oxidised to form the N-oxide metabolite. Both parent substance and metabolites are partly excreted as glucuronides. After multiple dosing the mean concentrations of the demethyl and didemethyl metabolites are usually 28-31% and <5%, respectively, of the escitalopram concentration. Biotransformation of escitalopram to the demethylated metabolite is mediated primarily by CYP2C19. Some contribution by the enzymes CYP3A4 and CYP2D6 is possible. The major metabolites have a significantly longer half-life than the parent drug.Escitalopram and major metabolites are assumed to be eliminated by both hepatic and renal routes, with the major part of the dose excreted as metabolites in the urine.
References
[1] sánchez c1, bergqvist pb, brennum lt, gupta s, hogg s, larsen a, wiborg o. escitalopram, the s-(+)-enantiomer of citalopram, is a selective serotonin reuptake inhibitor with potent effects in animal models predictive of antidepressant and anxiolytic activities. psychopharmacology (berl). 2003 jun;167(4):353-62. epub 2003 apr 26.
Properties of Escitalopram
| Boiling point: | 428.3±45.0 °C(Predicted) |
| alpha | D +12.33° (c = 1 in methanol) |
| Density | 1.18±0.1 g/cm3(Predicted) |
| storage temp. | 2-8°C |
| solubility | Soluble in DMSO |
| form | Powder |
| pka | 9.57±0.28(Predicted) |
| color | Off-white to light yellow |
| CAS DataBase Reference | 128196-01-0(CAS DataBase Reference) |
Safety information for Escitalopram
Computed Descriptors for Escitalopram
New Products
4,4-Difluoropiperidine hydrochloride tert-butyl 9-methoxy-3-azaspiro[5.5]undecane-3-carboxylate Indole Methyl Resin N-Isopropylurea N,N-Dicyclohexylcarbodiimide(DCC) MELDRUMS ACID 5-METHYLISOXAZOLE-4-CARBOXYLIC ACID Magnessium Bis glycinate Zinc ascorbate 1-bromo-2-butyne 2-acetamidophenol 9(10H)-anthracenone Erythrosin B, 4-Piperidinopiperidine 2-((4-morpholinophenylamino) (methylthio) methylene) malononitrile 2,4-dihydroxybenzaldehyde 3-(4-morpholinophenylamino)-5-amino-1H-pyrazole-4-carbonitrile Methyl 2-methylquinoline-6-carboxylate 2,6-dichloro-4-nitropyridine 4-Bromo-2-chlorobenzonitrile 2-(benzylamino)acetic acid hydrochloride 4-(tert-Butoxycarbonylamino)but- 2-ynoic acid 3,4-dihydro-2H-benzo[b][1,4]dioxepine 1-Phenyl-1-cycloprppanecarboxylicacidRelated products of tetrahydrofuran
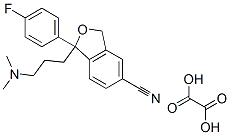

You may like
-
![128196-01-0 S-(+)-1-[3-(Dimethylamino)propyl]-1-(4-fluorophenyl)-1,3-dihydro-5-isobenzofurancarbonitrile oxalate 98%](https://img.chemicalbook.in//ProductImageIndia/2024-03/Raw/349de44d-af3d-46bd-9f4c-def49f31471c.png) 128196-01-0 S-(+)-1-[3-(Dimethylamino)propyl]-1-(4-fluorophenyl)-1,3-dihydro-5-isobenzofurancarbonitrile oxalate 98%View Details
128196-01-0 S-(+)-1-[3-(Dimethylamino)propyl]-1-(4-fluorophenyl)-1,3-dihydro-5-isobenzofurancarbonitrile oxalate 98%View Details
128196-01-0 -
 128196-01-0 98%View Details
128196-01-0 98%View Details
128196-01-0 -
Escitalopram 98%View Details
128196-01-0 -
 Escitalopram 128196-01-0 98%View Details
Escitalopram 128196-01-0 98%View Details
128196-01-0 -
 128196-01-0 Escitalopram 98%View Details
128196-01-0 Escitalopram 98%View Details
128196-01-0 -
128196-01-0 98%View Details
128196-01-0 -
 Escitalopram 98%View Details
Escitalopram 98%View Details
128196-01-0 -
 128196-01-0 Escitalopram 98%View Details
128196-01-0 Escitalopram 98%View Details
128196-01-0