
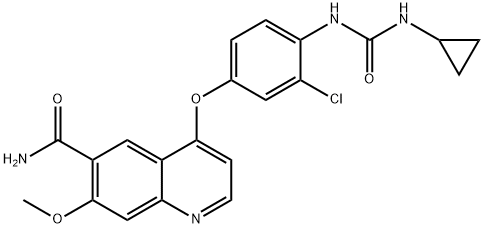
Trametinib
- CAS NO.:871700-17-3
- Empirical Formula: C26H23FIN5O4
- Molecular Weight: 615.39
- MDL number: MFCD17215075
- SAFETY DATA SHEET (SDS)
- Update Date: 2025-12-26 18:10:20
What is Trametinib?
Absorption
Following oral administration, trametinib is rapidly and readily absorbed. The absorption was examined in patients with solid tumours and BRAF V600 mutation-positive metastatic melanoma. Following the administration of trametinib tablets 0.125 mg (0.0625 times the approved recommended adult dosage) to 4 mg (2 times the approved recommended adult dosage) daily, both Cmax and AUC increased dose-proportionally. Intersubject variability in AUC and Cmax at steady state is 22% and 28%, respectively. Trametinib accumulates with daily repeat dosing with a mean accumulation ratio of 6.0 at 2 mg once daily dose. Steady-state was achieved by Day 15.
The mean absolute bioavailability of trametinib is 72% for oral tablets and 81% for oral solution. The Tmax is 1.5 hours. A high-fat, high-calorie meal (approximately 1000 calories) decreased trametinib AUC by 24% and Cmax by 70%, and delayed Tmax by approximately four hours as compared with fasted conditions.
Toxicity
There is no information regarding the acute toxicity (LD50) of trametinib. The highest doses of trametinib evaluated in clinical trials were 4 mg orally once daily and 10 mg administered orally once daily on two consecutive days, followed by 3 mg once daily. Out of seven patients treated on one of these two schedules, two patients experienced retinal pigment epithelial detachments. In clinical trials with trametinib monotherapy, one case of accidental overdose was reported from a single dose of 4 mg: no adverse events were reported in this event. Since trametinib is highly bound to plasma proteins, hemodialysis is likely to be ineffective in the treatment of drug overdose.
Description
Trametinib is an inhibitor of MEK1 and -2. It inhibits B-RAF- and C-RAF-induced phosphorylation of MEK1 (IC50s = 3.4 and 1.8 nM, respectively) and MEK2 (IC50s = 1.6 and 0.92 nM, respectively). Trametinib inhibits the growth of two human colorectal cancer cell lines expressing mutant B-RAF (IC50s = 0.48 and 0.52 nM) and seven cell lines expressing mutant K-Ras (IC50s = 2.2-174 nM) but does not inhibit the growth of wild-type COLO 320DM cells expressing both B-RAF and K-Ras (IC50 = >10,000 nM). It reduces tumor growth in HT-29 and COLO 205 mouse xenograft models when used at doses of 0.3 and 1 mg/kg per day. Trametinib (0.03 and 0.1 mg/kg per day) also decreases M. tuberculosis-induced increases in hind paw volume in a rat model of arthritis. Formulations containing trametinib, in combination with dabrafenib, have been used in the treatment of metastatic mutant B-RAFV600E melanoma.
The Uses of Trametinib
Used in the systematic treatment of advanced cutaneous melanoma. An EGFR kinase inhibitor.
The Uses of Trametinib
antiprotozoal
The Uses of Trametinib
Used in the systematic treatment of advanced cutaneous melanoma. An EGFR kinase inhibitor and potent MEK inhibitor.
Background
Trametinib is an orally bioavailable mitogen-activated extracellular signal-regulated kinase 1 (MEK1) and MEK2 inhibitor. It was first approved by the FDA in May 2013 for the treatment of melanoma. It was later approved by Health Canada on July 18, 2013 and by the European Commission on June 30, 2014. Trametinib is currently approved to treat a variety of cancers with BRAF mutations, such as non-small cell lung cancer and thyroid cancer, as monotherapy or in combination with dabrafenib, a BRAF inhibitor, for improved therapeutic efficacy. Originally developed by Japan Tobacco, trametinib was initially investigated for treating inflammation, but further studies for this indication were not pursued.
Indications
Trametinib is indicated as monotherapy for the treatment of BRAF-inhibitor treatment-na?ve patients with unresectable or metastatic melanoma with BRAF V600E or V600K mutations.
It is used in combination with dabrafenib for the:
In the US, BRAF V600E or V600K mutations must be detected by an FDA-approved test. Trametinib is not indicated for the treatment of patients with colorectal cancer because of known intrinsic resistance to BRAF inhibition.
What are the applications of Application
Trametinib is an allosteric inhibitor of MEK1/MEK2 that reduces tumor growth in mouse xenograft models.
Definition
ChEBI: A pyridopyrimidine that is used (as its dimethyl sulfoxide addition compound) for the treatment of patients with unresectable or metastatic melanoma with BRAF V600E or V600K mutations, and who have not received prior BRAF inhibitor treatment.
Indications
MEK, also known as MAPK, is a dual specificity threonine/tyrosine kinase that is a key node in the Raf–Ras–MEK signaling pathway. Small-molecule MEK inhibitors represent the largest group of type III allosteric inhibitors that do not bind to the ATP binding pocket. As of December 2016, besides the FDA-approved MEK1/2 inhibitors trametinib (Mekinist(R), GlaxoSmithKline) and cobimetinib (Cotellic(R), Roche), over 10 MEK inhibitors are currently in clinical trials. Trametinib was approved by FDA in 2013 for the treatment of patients with either B-Raf V600E or V600K mutated metastatic melanoma. Considering the fact that MEK and Raf are different kinases along the same pathway of Ras–Raf–MEK/ERK signaling cascade, combination strategies using both MEK and B-Raf inhibitors were utilized to overcome the observed progression using single-agent trametinib, which usually occurs within 7months. FDA approved the combination of trametinib and dabrafenib for the treatment of B-Raf V600E/K mutated metastatic melanoma in January 2014 and the combination of cobimetinib and vemurafenib for the same type of indication. Although significant improvement in progression-free survival was observed using MEK/B-Raf combination strategy, the incidence of some common adverse effects, such as vomiting, diarrhea, nausea, rash, and pyrexia, also increased.
brand name
Mekinist
General Description
Class: dual threonine/tyrosine kinase; Treatment: melanoma with BRAF mutations; Other name: JTP-74057, GSK1120212; Oral bioavailability = 72%; Elimination half-life = 10 days; Protein binding = 97.4%
Pharmacokinetics
Trametinib inhibits cell growth of various BRAF V600 mutation-positive tumours in vitro and in vivo. Trametinib is often used in combination with dabrafenib, a BRAF inhibitor. In BRAF-mutant colorectal cancer, induction of EGFR-mediated MAPK pathway re-activation has been identified as a mechanism of intrinsic resistance to BRAF inhibitors.
Pharmacokinetics
Trametinib has favorable pharmacokinetic
properties: quick oral absorption with tmax of 0.5–1.5
h, long duration of action with effective t1/2 of 4 days,
and elimination t1/2 of 10 days, as well as good oral
bioavailability (72%). The unusual long half-life and
good potency contribute a once-daily administration
of just 2 mg, the lowest dosage of all the MEK
inhibitors now in use (Table 2). Trametinib is metabolized predominantly via
deacetylation to give 6, which subsequently
undergoes hydroxylation to give 8 or glucuronidation
to afford 7 (Fig. 4).

Clinical Use
Trametinib (GSK1120212) is an oral MEK inhibitor which has demonstrated excellent results in combination therapy for BRAF-mutated melanoma and is FDA approved in combination with BRAF inhibitors for that indication. Trametinib was evaluated initially as a single agent in KRAS mutant NSCLC in comparison with docetaxel and pemetrexed. Results as a single agent were not impressive, with an ORR of only 12% in these patients.
Drug interactions
Potentially hazardous interactions with other drugs
Antipsychotics: avoid with clozapine - increased risk
of agranulocytosis.
Metabolism
Trametinib predominantly undergoes deacetylation mediated by carboxylesterases (i.e., carboxylesterase 1b/c and 2) and other hydrolytic enzymes. The deacetylated metabolite may further be glucuronidated. In vitro findings suggest that deacetylation may also be accompanied by mono-oxygenation, hydroxylation, and glucuronidation. CYP3A4-mediated oxidation is a minor pathway. Four metabolites (M1/2/3/4) have been characterized in patients with advanced cancers. In vitro, the M1 and M3 metabolites demonstrated approximately equal or 10-fold less potent phospho-MEK1-inhibiting activity than the parent compound.
Following a single dose of [14C]-trametinib, approximately 50% of circulating radioactivity represented the parent compound. According to findings from metabolite profiling after repeat dosing of trametinib, unchanged parent drug accounted for greater than or equal to 75% of drug-related material in plasma.
Metabolism
Metabolised mainly by deacetylation alone or in
combination with mono-oxygenation. The deacetylated
metabolite was further metabolised by glucuronidation.
CYP3A4 oxidation is considered a minor pathway
of metabolism. The deacetylation is mediated by
the carboxyl-esterases 1b, 1c and 2, with possible
contributions by other hydrolytic enzymes.
Total dose recovery was low after a 10-day collection
period (<50
%) following administration of a single
oral dose of radiolabelled trametinib, due to the long
elimination
Half-life. Trametinib was excreted mainly
in the faeces (>80
% of recovered radioactivity) and to a
minor extent in urine (≤19
%).
Storage
Store at -20°C
References
1) Gilmartin?et al.?(2011),?GSK1120212 (JTP-74057) is an Inhibitor of MEK Activity and Activation with Favorable Pharmacokinetic Properties for Sustained In Vivo Pathway Inhibition; Clin. Cancer Res.?17?989 2) Abe?et al. (2011),?Discovery of a Highly Potent and Selective MEK Inhibitor: GSK1120212 (JTP-74057 DMSO solvate); ACS Med. Chem. Lett.?2?320 3) Allegrezza?et al.?(2016),?Trametinib Drives T-cell-Dependent Control of KRAS Tumors by Inhibiting Pathological Myelopoiesis; Cancer Res.?76?6253 4) Vella?et al.?(2014),?MEK inhibition, alone or in combination with BRAF inhibition, affects multiple functions of isolated normal human lymphocytes and dendritic cells; Cancer Immunol. Res.?2?351 5) Yamaguchi?et al.?(2012),?Suppressive effect of an orally active MEK1/2 inhibitor in two different animal models for rheumatoid arthritis: a comparison with leflunomide; Inflamm. Res.?61?445
Properties of Trametinib
| Melting point: | 300-301 °C |
| Density | 1.74 |
| storage temp. | -20°C |
| solubility | Soluble in DMSO (up to 20 mg/ml) |
| form | White solid. |
| pka | 14.76±0.70(Predicted) |
| color | White |
| Stability: | Stable for 1 year from date of purchase as supplied. Solutions in DMSO may be stored at -20°C for up to 2 months. |
Safety information for Trametinib
Computed Descriptors for Trametinib
Trametinib manufacturer
New Products
4,4-Difluoropiperidine hydrochloride tert-butyl 9-methoxy-3-azaspiro[5.5]undecane-3-carboxylate Indole Methyl Resin N-Isopropylurea N,N-Dicyclohexylcarbodiimide(DCC) MELDRUMS ACID 5-METHYLISOXAZOLE-4-CARBOXYLIC ACID Magnessium Bis glycinate Zinc ascorbate 1-bromo-2-butyne 2-acetamidophenol 9(10H)-anthracenone Erythrosin B, 4-Piperidinopiperidine 2-((4-morpholinophenylamino) (methylthio) methylene) malononitrile 2,4-dihydroxybenzaldehyde 3-(4-morpholinophenylamino)-5-amino-1H-pyrazole-4-carbonitrile Methyl 2-methylquinoline-6-carboxylate 2,6-dichloro-4-nitropyridine 4-Bromo-2-chlorobenzonitrile 2-(benzylamino)acetic acid hydrochloride 4-(tert-Butoxycarbonylamino)but- 2-ynoic acid 3,4-dihydro-2H-benzo[b][1,4]dioxepine 1-Phenyl-1-cycloprppanecarboxylicacidRelated products of tetrahydrofuran
You may like
-
 Trametinib 99% (HPLC) CAS 871700-17-3View Details
Trametinib 99% (HPLC) CAS 871700-17-3View Details
871700-17-3 -
 Luci-Tram 2mg Trametinib Tablet, Melanoma, Skin CancerView Details
Luci-Tram 2mg Trametinib Tablet, Melanoma, Skin CancerView Details
871700-17-3 -
 Trametinib 0 .5 mg tabletView Details
Trametinib 0 .5 mg tabletView Details
871700-17-3 -
 Trametinib 0 .5 mg tabletView Details
Trametinib 0 .5 mg tabletView Details
871700-17-3 -
3-(4-amino-1-oxoisoindolin-2-yl)-1-methylpiperidine-2,6-dione 98%View Details
-
20677-73-0 (2,2-diethoxyethyl)methylamine 98%View Details
20677-73-0 -
3-(4-(hydroxyamino)-1-oxoisoindolin-2-yl)piperidine-2,6-dione 98%View Details
-
57381-49-4 2-bromo-4-chlorobenzonitrile 98%View Details
57381-49-4