Darunavir
Synonym(s):[(1R,5S,6R)-2,8-dioxabicyclo[3.3.0]oct-6-yl] N-[(2S,3R)-4- [(4-aminophenyl)sulfonyl- (2-methylpropyl)amino]-3-hydroxy-1-phenyl- butan-2-yl];TMC-114;UIC-94017
- CAS NO.:206361-99-1
- Empirical Formula: C27H37N3O7S
- Molecular Weight: 547.66
- MDL number: MFCD09260006
- EINECS: 606-590-1
- SAFETY DATA SHEET (SDS)
- Update Date: 2024-03-26 17:16:27
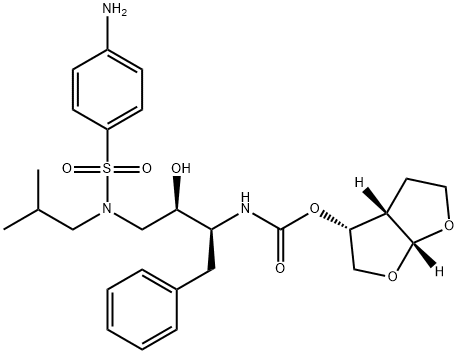
What is Darunavir?
Absorption
The absolute oral bioavailability of one single 600 mg dose of darunavir alone and with 100 mg of ritonavir twice a day was 37% and 82%, respectively. Exposure to darunavir in boosted patients has been found to be 11 times higher than in unboosted patients. Tmax is achieved approximately 2.4 to 4 hours after oral administration.
When darunavir is taken with food, the Cmax and AUC of darunavir given with ritonavir increase by 30% when compared to the fasted state.
Toxicity
LD50 information for darunavir is not readily available in the literature. One-time doses of up to 3,200 mg of darunavir in an oral solution and up to 1,600 mg of the tablet formulation of darunavir with ritonavir have been given volunteers without significant symptoms.
Information about an overdose with darunavir with ritonavir is limited. No specific antidote exists for this drug. Treatment of In the case of an overdose, employ general supportive measures. Monitor vital signs and clinical status. It is unlikely that darunavir not amenable to removal by dialysis due to its high level of protein binding.
Description
Darunavir is the latest weapon in the arsenal of agents to combat human immunodeficiency virus type 1(HIV-1). As an HIV-1 protease inhibitor, its mechanism of action involves blocking the cleavage of the gag and gag–pol polyproteins into functional proteins essential for the production of infectious progeny virus; the result is the production of immature, noninfectious viral particles. Compared to predecessor HIV protease inhibitors, darunavir retains activity against resistant stains, a critical factor with the continual emergence of multidrug- resistant (MDR) mutants. Despite experiencing a 13-fold reduction in binding to MDR HIV-1 protease, this binding is 1.5 orders of magnitude tighter than the first-generation protease inhibitors. Furthermore, darunavir exhibits less than a 10-fold decrease in susceptibility in cell culture against 90% of 3309 clinical isolates resistant to amprenavir, atazanavir, indinavir, lopinavir, nelfinavir, ritonavir, saquinavir, and tipranavir. In contrast, darunavir-resistant viruses display limited susceptibility to only tipranavir, suggesting limited cross-resistance between these two protease inhibitors. To avoid the issues of the peptide-based protease inhibitors, darunavir has evolved from a structure-based design effort to minimize peptidic features and reduce molecular weight and complexity.
Chemical properties
White Amorphous Solid
Originator
Tibotec/J&J (US)
The Uses of Darunavir
Second-generation HIV-1-protease inhibitor; structurally similar to amprenavir. It is an antiviral agent and a COVID-19-related research product.
Definition
ChEBI: An N,N-disubstituted benzenesulfonamide bearing an unsubstituted amino group at the 4-position, used for the treatment of HIV infection. A second-generation HIV protease inhibitor, darunavir was designed to form robust interactions with the protease enzyme from many strains of HIV, including those from treatment-experienced patients with multiple resistance mutations to other protease inhibitors.
Indications
Darunavir, co-administered with ritonavir, and with other antiretroviral agents, is indicated for the treatment of human immunodeficiency virus (HIV) in children age 3 or above and adults with HIV-1 infection.
Background
Darunavir is a protease inhibitor used with other HIV protease inhibitor drugs as well as ritonavir for the effective management of HIV-1 infection. As a second-generation protease inhibitor, darunavir is designed to combat resistance to standard HIV therapy. It was initially approved by the FDA in 2006.
Darunavir is being studied as a possible treatment for SARS-CoV-2, the coronavirus responsible for COVID-19, due to in vitro evidence supporting its ability to combat this infection. Clinical trials are underway and are expected to conclude in August 2020.
brand name
Prezista
Acquired resistance
Darunavir is less affected than other protease inhibitors by mutations to resistance, but subgroups with more than 10 cumulative mutations show a >10-fold (median value) decrease in susceptibility. The major resistance mutations occur at positions 50 (150V), 54 (I50M/L), 76 (L76V) and 84 (I84V) of the protease gene.
Pharmaceutical Applications
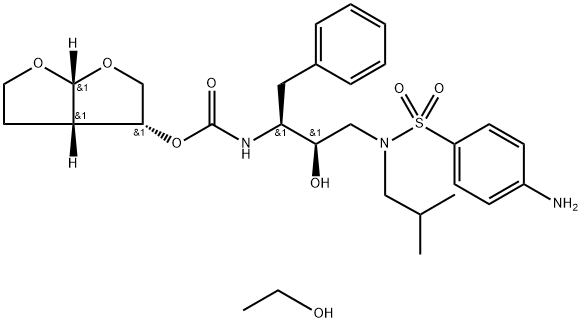
A synthetic compound formulated as the ethanolate for oral use in combination with ritonavir.
Biochem/physiol Actions
Darunavir has been sanctioned by the food and drug administration (FDA) as the first treatment of drug-resistant human immunodeficiency virus (HIV).
Pharmacokinetics
Darunavir is an inhibitor of the human immunodeficiency virus (HIV) protease, which prevents HIV viral replication. When administered with ritonavir in combination antiretroviral therapy, darunavir significantly decreases viral load and increases CD4 cell counts, decreasing the morbidity and mortality of HIV infection.
Pharmacokinetics
Oral absorption: c. 82%
Cmax 600 mg once daily + ritonavir 100
mg twice daily:
c. 6500 μg/L
Cmin 600 mg oral + ritonavir
100 mg twice daily:
c. 3578 μg/L
Plasma half-life: c. 15 h
Volume of distribution: c. 131 L
Plasma protein binding: c. 95%
A single 600 mg dose given orally in combination with ritonavir
100 mg every 12 h increased the systemic exposure of
darunavir approximately 14-fold. The relative bioavailability
is 30% lower when administered with food in the presence of
low-dose ritonavir. Distribution into human CSF, semen or
breast milk has not yet been determined.
At least three oxidative metabolites, mediated predominantly
through CYP3A4, have been identified in humans;
all are at least 10-fold less active than the parent compound
against HIV. Around 80% and 14% of the dose is found in the
feces and urine, respectively. It should be used with caution
in mild–moderate hepatic impairment and avoided in patients
with more severe impairment.
Clinical Use
Treatment of HIV infection (in combination with other antiretroviral drugs)
Side Effects
In phase III studies the most common adverse events were diarrhea, nausea, headache and nasopharyngitis. Patients coinfected with hepatitis B or C did not have a higher incidence of adverse events.
Synthesis
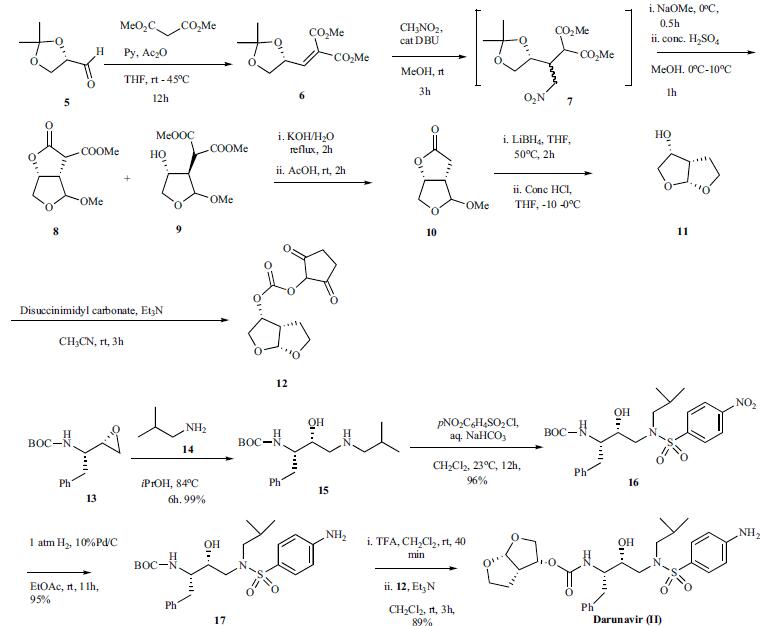
Several routes to the synthesis of darunavir have been reported utilizing the chiral hexahydro-furo[2,3-b]furan-3-ol carbonate 12 and several chiral syntheses of bisfuranol 12 have been disclosed as well. One route that has been performed on kilogram scale is highlighted in the scheme. Thus 2,3-Oisopropylidene- glyceraldehyde 5 was stirred with dimethyl malonate at RT for 3 h in tetrahydrofuran followed by addition of pyridine and heating to 45oC. Then acetic anhydride was added at 45oC over 4h and stirred at that temperature for 12 h. Concentration of the reaction followed by basic workup and extraction with toluene and solvent swap to methanol gave the products as a 23.6% solution in methanol. Nitromethane was added to this methanol solution followed by the addition of DBU over 30 min keeping the reaction temperature below 25oC. Stirring the reaction for an additional 3 h afforded intermediate 7. The reaction was cooled to 0oC and sodium methoxide was added dropwise over 30 min After stirring the reaction for 30 min, the reaction was added slowly over 1h to conc. H2SO4 in methanol at 0oC while ensuring the temperature did not exceed 10oC. This cooled reaction mixture (0oC) was then added to a vigorously stirred mixture of ethyl acetate and 1N sodium hydrogen carbonate at 0oC. The organic layer was separated, washed with brine and concentrated to give the residue containing a mixture of 8 and 9. This mixture was dissolved in methanol then water and potassium hydroxide were added and the resulting mixture was heated at reflux for 2 h. The reaction was cooled to 35oC and acetic acid was added and the resulting mixture concentrated. Additional acetic acid was added and stirred at room temperature for 2 h. The mixture was concentrated, diluted with water and extracted with ethylacetate. The ethylacetate layer was washed with 1N sodium bicarbonate three times and the organic layer was concentrated and diluted with isopropanol. The isopropanol mixture was then heated to 60-70oC and further evaporation of isopropanol under reduced pressure to a concentrated volume with cooling to 0oC over 4-5 h, allowed for the crystallization of product 10. After filtration and drying, the intermediate lactone 10 was dissolved in THF and treated over 30 min with a solution of lithium borohydride in THF. The reaction was warmed to 50oC over 1 h and stirred at that temperature for 2h. The resulting suspension was cooled to -10oC and conc. HCl was added slowly over 4h, while maintaining the temperature below 0oC. Solvent swap was done by concentrating to a small volume and addition of ethyl acetate and further concentration of the solvent with continuous addition of ethylacetate. Following this procedure, when the final ratio of THF:ethylacetate reached 4:1 ratio, the mixture was cooled to 0oC and filtered off while washing the filter cake with more ethylacetate. Concentration of the filtrate to dryness gave the hexahydro-furo [2,3-b]furan-3-ol 11 which was confirmed by NMR and chiral gas chromatography. Carbonate intermediate 12 was prepared in 66% yield by treating 11 with disuccinimidyl carbonate at RT for 3h in the presence of triethylamine. Since the process scale synthesis of darunavir has not been disclosed, the latest reported synthesis is highlighted. The commercially available epoxide 13 was mixed with isobutyl amine in isopropanol at RT and refluxed for 6h. The reaction was concentrated and purified by chromatography to provide amine 15 (99%). p-Nitrophenyl sulfonyl chloride was added to a mixture of the amine 15 in dichloromethane and saturated aqueous bicarbonate at RT and stirred for 12 h to give sulfonamide 16 in 96% yield after purification. Hydrogenation of 16 with 10% Pd/C under 1 atm hydrogen for 11h at room temperature gave aniline 17 in 95% yield. The BOC group was removed by treating 17 with TFA in dichloromethane and the resulting amine was reacted with carbonate 12 in the presence of triethylamine for 3h to provide the desired darunavir (II) in 89% yield.
Drug interactions
Potentially hazardous interactions with other drugs
Antibacterials: rifabutin concentration increased -
reduce dose of rifabutin; darunavir concentration
reduced by rifampicin - avoid.
Anticoagulants: avoid with apixaban and rivaroxaban
Antidepressants: possibly reduced concentration of
paroxetine and sertraline; darunavir concentration
reduced by St John’s wort - avoid.
Antimalarials: concentration of lumefantrine increased;
possibly increases concentration of quinine
Antipsychotics: possibly increases concentration of
aripiprazole (reduce dose of aripiprazole); possibly
increases quetiapine concentration - avoid.
Antivirals: avoid with boceprevir or telaprevir; take
didanosine 1 hour before or 2 hours after darunavir
administration; concentration reduced by efavirenz
- adjust dose; concentration of both drugs increased
with indinavir and simeprevir - avoid with simeprevir;
concentration reduced by lopinavir, also concentration
of lopinavir increased - avoid; concentration of
maraviroc increased, consider reducing dose of
maraviroc; concentration of paritaprevir increased
and paritaprevir reduces darunavir concentration;
concentration reduced by saquinavir; increased risk of
rash with raltegravir; avoid with telaprevir.
Cytotoxics: possibly increases bosutinib concentration,
avoid or reduce dose of bosutinib; possibly increases
everolimus concentration - avoid; possibly increases
ibrutinib concentration - reduce ibrutinib dose.
Ergot alkaloids: increased risk of ergotism - avoid.
Lipid-lowering drugs: possibly increased risk of
myopathy with atorvastatin and rosuvastatin, reduce
dose of rosuvastatin; possibly increases pravastatin
concentration; avoid with lomitapide; avoid with
simvastatin.1
Orlistat: absorption of darunavir possibly reduced.
Ranolazine: possibly increases ranolazine
concentration - avoid.
Mechanism of action
Darunavir inhibits the HIV protease enzyme by forming an inhibitor-enzyme complex thereby preventing cleavage of the gag-pol polyproteins. Immature, non-infectious viral particles are subsequently produced.
Metabolism
Darunavir is metabolized by oxidation by the cytochrome P450 system (mainly the isoenzyme CYP3A4), with at least 3 metabolites showing some antiretroviral activity. About 80% of a dose is excreted in the faeces, with 41.2% of this as an unchanged drug; 14% is excreted in the urine. Darunavir is extensively metabolized in subjects who do not receive a booster, primarily via carbamate hydrolysis, isobutyl aliphatic hydroxylation, and aniline aromatic hydroxylation, as well as both benzylic aromatic hydroxylation and glucuronidation.
storage
Store at -20°C
References
Koh et al. (2003), Novel bis-tetrahydrofuranylurethane-containing nonpeptidic protease inhibitor (PI UIC-94017 (YMC114) with potent activity against multi-PI-resistant human immunodeficiency virus in vitro; Antimicrob. Agents Chemother., 47 2123 Surleraux et al. (2005), Discovery and selection of TMC114, a next generation HIV-1 protease inhibitor; J. Med. Chem., 48 1813 Beck et al. (2020), Predicting commercially available antiviral drugs that may act on the novel coronavirus (SARS-CoV-2) through a drug-target interaction deep learning model; Comput. Struct. Biotechnol. J, 18 784 Khan et al. (2020), Identification of chymotrypsin-like protease inhibitors of SARS-VCoV-2 via integrated computational approach; J. Biomol.Struct. Dyn., 39 2607 De Meyers et al. (2020), Lack of antiviral activity of darunavir against SARS-CoV-2; J. Infect. Dis, 97 7 Kim et al. (2020), Use of Darunavir-Cobicistat as a Treatment Option for Critically Ill Patients with SARS-CoV-2 Infection; Yonsei Med. J., 61 826
Properties of Darunavir
| Melting point: | 74-760C |
| Density | 1.34±0.1 g/cm3(Predicted) |
| storage temp. | -20°C |
| solubility | Soluble in DMSO (>25 mg/ml) |
| pka | 11.43±0.46(Predicted) |
| form | powder |
| color | white to beige |
| Stability: | Stable for 1 year from date of purchase as supplied. Solutions in DMSO may be stored at -20°C for up to 3 months. |
Safety information for Darunavir
| Signal word | Warning |
| Pictogram(s) |
 Exclamation Mark Irritant GHS07 |
| GHS Hazard Statements |
H302:Acute toxicity,oral H315:Skin corrosion/irritation H319:Serious eye damage/eye irritation H335:Specific target organ toxicity, single exposure;Respiratory tract irritation |
| Precautionary Statement Codes |
P261:Avoid breathing dust/fume/gas/mist/vapours/spray. P305+P351+P338:IF IN EYES: Rinse cautiously with water for several minutes. Remove contact lenses, if present and easy to do. Continuerinsing. |
Computed Descriptors for Darunavir
Darunavir manufacturer
HRV Global Life Sciences
VIVIN DRUGS AND PHARMACEUTICALS LTD
New Products
(S)-3-Aminobutanenitrile hydrochloride 4-Methylphenylacetic acid N-Boc-D-alaninol N-BOC-D/L-ALANINOL Tert-butyl bis(2-chloroethyl)carbamate 3-Morpholino-1-(4-nitrophenyl)-5,6-dihydropyridin- 2(1H)-one Furan-2,5-Dicarboxylic Acid Tropic acid 1-Bromo-3,5-Di-Tert-Butylbenzene S-2-CHLORO PROPIONIC ACID ETHYL ISOCYANOACETATE 2-Bromo-1,3-Bis(Dimethylamino)Trimethinium Hexafluorophosphate 4-IODO BENZOIC ACID 3-NITRO-2-METHYL ANILINE 1-(2,4-DICHLOROPHENYL) ETHANAMINE (2-Hydroxyphenyl)acetonitrile 4-Bromopyrazole 2-(Cyanocyclohexyl)acetic acid 4-methoxy-3,5-dinitropyridine 1-(4-(aminomethyl)benzyl)urea hydrochloride 2-aminopropyl benzoate hydrochloride diethyl 2-(2-((tertbutoxycarbonyl)amino) ethyl)malonate tert-butyl 4- (ureidomethyl)benzylcarbamate Ethyl-2-chloro((4-methoxyphenyl)hydrazono)acetateRelated products of tetrahydrofuran
![4-AMINO-N-[(2R,3S)-3-AMINO-2-HYDROXY-4-PHENYLBUTYL]-N-ISOBUTYLBENZENE-1-SULFONAMIDE](https://img.chemicalbook.in/CAS/GIF/169280-56-2.gif)
![(3R,3αS,6αR)-Hexahydrofuro[2,3-β]furan-3-yl-4-nitrophenyl carbonate](https://img.chemicalbook.in/CAS/20180808/GIF/252873-35-1.gif)
![tert-Butyl [(1S,2R)-1-benzyl-2-hydroxy-3-[isobutyl[(4-nitrophenyl)sulfonyl]amino]propyl]carbamate](https://img.chemicalbook.in/CAS/GIF/191226-98-9.gif)
![[(3R,3aS,6aR)-Hydroxyhexahydrofuro[2,3-β]furanyl Succinimidyl Carbonate](https://img.chemicalbook.in/CAS/GIF/253265-97-3.gif)
You may like
-
 206361-99-1 Darunavir 98%View Details
206361-99-1 Darunavir 98%View Details
206361-99-1 -
 206361-99-1 98%View Details
206361-99-1 98%View Details
206361-99-1 -
 Darunavir 99%View Details
Darunavir 99%View Details -
 Daruvir 98%View Details
Daruvir 98%View Details
206361-99-1 -
 Darunavir 98%View Details
Darunavir 98%View Details -
 Darunavir 98%View Details
Darunavir 98%View Details -
206361-99-1 Darunavir 95-99 %View Details
206361-99-1 -
Darunavir CAS 206361-99-1View Details
206361-99-1