
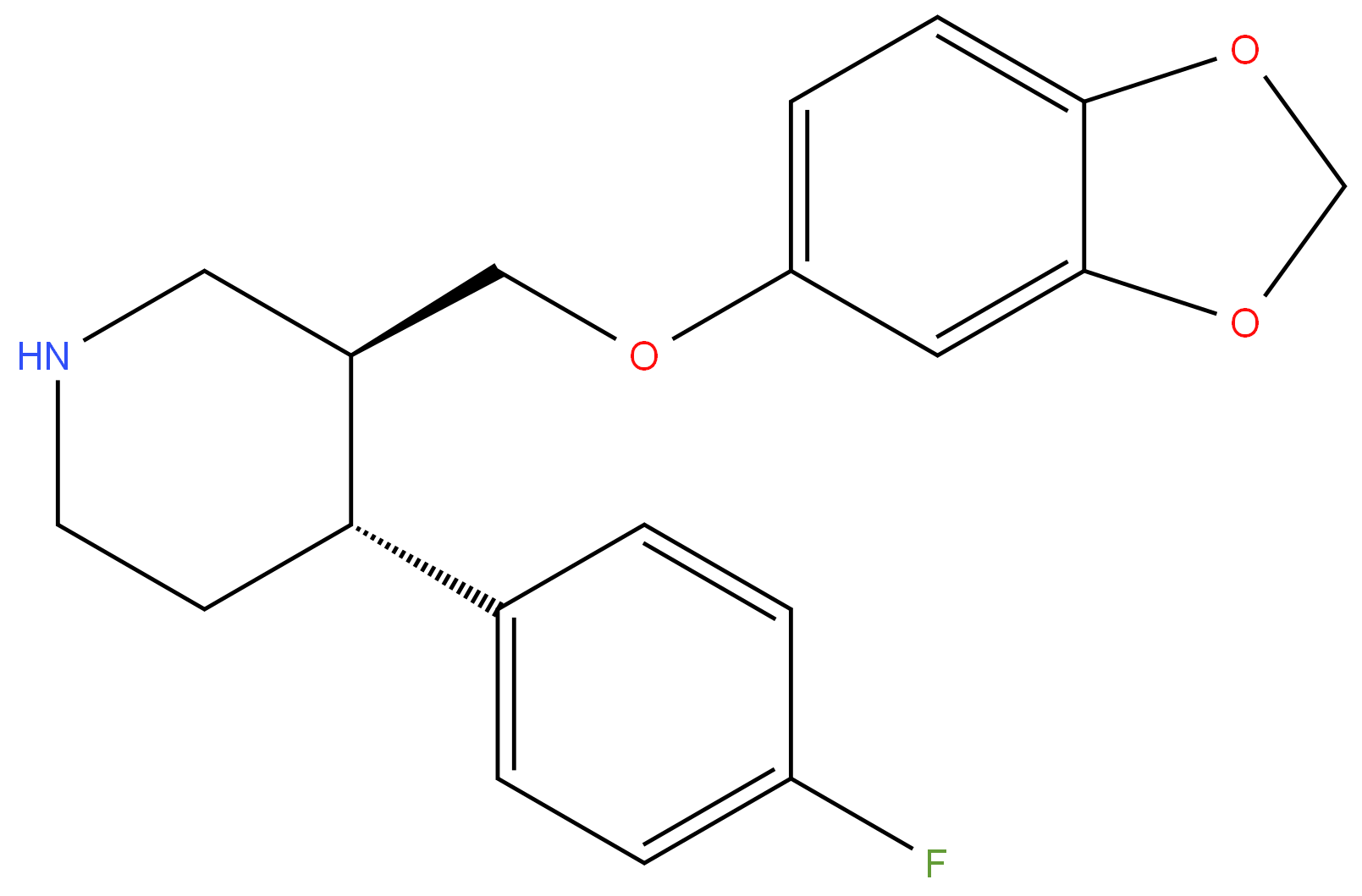
Paroxetine
- CAS NO.:61869-08-7
- Empirical Formula: C19H20FNO3
- Molecular Weight: 329.37
- MDL number: MFCD00869588
- EINECS: 682-717-4
- SAFETY DATA SHEET (SDS)
- Update Date: 2025-12-29 14:06:09

What is Paroxetine?
Absorption
Paroxetine is readily absorbed from the gastrointestinal tract. Due to the first-pass metabolism, the bioavailability ranges from 30-60%. Cmax is attained 2 to 8 hours after an oral dose. Mean Tmax is 4.3 hours in healthy patients. The steady-state concentration of paroxetine is achieved within 7 to 14 days of oral therapy. In a pharmacokinetic study, AUC in healthy patients was 574 ng·h/mL and 1053 ng·h/mL in those with moderate renal impairment.
Toxicity
The acute LD50 in mice and rats is 350 mg/kg.
Overdose information
The lowest dose of paroxetine reported to lead to a fatal outcome is approximately 400 mg. The largest reported paroxetine overdose from which a patient has survived and recovered is a dose of 2000 mg. Common manifestations in a paroxetine overdose include fatigue, fever, insomnia hypertension, tachycardia, nausea, vomiting, somnolence, tremor, dizziness, agitation, confusion, anxious symptoms, headache, insomnia, hyperhidrosis, dilated pupils, seizures, paresthesia, serotonin syndrome, involuntary muscle contraction, and change in mental status. It should be noted that in some cases, patients may have consumed alcohol in addition to taking an overdose of paroxetine. Some of these symptoms may also be seen with clinical use. There is no specific antidote to an overdose of paroxetine.
Description
Paroxetine is a new highly selective serotonin reuptake inhibitor, mechanistically similar to fluoxetine, fluvoxamine and sertraline, introduced for the treatment of all types of depressive illnesses including depression associated with anxiety. It is reportedly non-sedating and non-stimulatory and compared to fluoxetine has a shorter duration of action (half-life of 24 hours versus 2 to 3 days). Paroxetine is also being investigated as a treatment for obesity, alcoholism and obsessive-compulsive disorders.
Description
Paroxetine (trade name Paxil) is an antidepressant drug in the selective serotonin reuptake inhibitor class. It was first marketed in 1992 by the company now known as GlaxoSmithKline. It has some significant side effects, and its sales have dropped off in recent years. A recent?report?describes a modification of paroxetine to prevent the inactivation of a liver enzyme.
Chemical properties
White Solid
Originator
As Ferrosan (Novo-Nordisk) (Denmark)
The Uses of Paroxetine
A istopically labelled selective serotonin reuptake inhibitor. Used as an antidepressant
Background
Paroxetine is a selective serotonin reuptake inhibitor (SSRI) drug commonly known as Paxil. It has a variety of uses, including the treatment of anxiety disorders, major depression, posttraumatic stress disorder, and symptoms of menopause, among others. It was approved by the FDA in the early 1990s and marketed by SmithKline Beecham. A unique feature of this drug is that it is highly potent and selective in its inhibition of serotonin reuptake and has little effect on other neurotransmitters. Because of its potent inhibition of serotonin reuptake, paroxetine is more likely to cause withdrawal effects upon cessation. Paroxetine is well tolerated in most patients with a similar adverse effect profile to other members of its drug class. The controlled release formulation was designed to decrease the likelihood of nausea that is sometimes associated with paroxetine.
Indications
Paroxetine is indicated for the management of depression, obsessive-compulsive disorder, panic disorder, social anxiety disorder, generalized anxiety disorder, posttraumatic stress disorder. One form of paroxetine, commercially known as Brisdelle, is used to manage mild to moderate vasomotor symptoms of menopause. Off-label, paroxetine may be used for the treatment of premature ejaculation or irritable bowel syndrome (IBS).
Definition
ChEBI: Paroxetine is a benzodioxole that consists of piperidine bearing 1,3-benzodioxol-5-yloxy)methyl and 4-fluorophenyl substituents at positions 3 and 4 respectively; the (3S,4R)-diastereomer. Highly potent and selective 5-HT uptake inhibitor that binds with high affinity to the serotonin transporter (Ki = 0.05 nM). Ki values are 1.1, 350 and 1100 nM for inhibition of [3H]-5-HT, [3H]-l-NA and [3H]-DA uptake respectively. Displays minimal affinity for alpha1-, alpha2- or beta-adrenoceptors, 5-HT2A, 5-HT1A, D2 or H1 receptors at concentrations below 1000 nM, however displays weak affinity for muscarinic ACh receptors (Ki = 42 nM). Antidepressant and anxiolytic in vivo. It has a role as an antidepressant, an anxiolytic drug, a serotonin uptake inhibitor, a hepatotoxic agent and a P450 inhibitor. It is a member of piperidines, a member of benzodioxoles, an organofluorine compound and an aromatic ether. It is functionally related to a monofluorobenzene. It is a conjugate base of a paroxetinium(1+).
Manufacturing Process
251 g of methyl-4-(4-fluorophenyl)-N-methyl-nipecotinate, 8 g of sodium methoxide and 500 ml benzene were refluxed for 2 h. The benzene solution was washed with cold water and evaporated to give the pure α-ester which was dissolved in a mixture of 320 ml of water and 450 ml concentrated hydrochloric acid. The solution was slowly distilled to remove methanol and finally evaporated to dryness in vacuo.
400 ml thionyl chloride were added in small portions to the solid. The mixture was allowed to stand for 3 h at room temperature and was then evaporated to dryness in vacuo with tetrachloroethane giving methyl-4-(4-fluorophenyl)-Nmethylnipecotic acid chloride. The acid chloride was added in small portions to a solution of 160 g (-)-menthol in 800 ml pyridine at a temperature of 0°-5°C. The mixture was allowed to stand at room temperature to the next day. Ice water and 50% sodium hydroxide were added, and the mixture was extracted with ether. The ether was dried with anhydrous magnesium sulphate, filtered and evaporated. Distillation in vacuo gave the menthol ester in a yield of 7580%. Boiling point at 0.05 mm Hg was 165°-170°C.
Racemic 4-(4-fluorophenyl)-1-methyl-1,2,3,6-tetrahydropyridine (50 g) was dissolved in a mixture of 21.6 ml of concentrated sulfuric acid and 50 ml of water. To the solution were added 25 ml of concentrated hydrochloric acid and 22.4 ml of 37% formaldehyde solution. The mixture was refluxed for 5 h, cooled, and 125 ml of concentrated ammonia were added. The mixture was extracted with 50 ml of toluene. Drying of the toluene solution and distillation gave 38 g of 4-(4-fluorophenyl)-3-hydroxymethyl-1-methyl-1,2,3,6tetrahydropyridine with boiling point 110°-120°C at 0.1 mm Hg.
13 g of the racemic compound and 22 g of (-)-dibenzoyltartaric acid were dissolved in 105 ml of hot methanol. On cooling, 9 g of salt of (-)-4-(4fluorophenyl)-3-hydroxymethyl-1-methyl-1,2,3,6-tetrahydropyridine crystallized. Melting point 167°-168°C.
38 g of (-)-4-(4-fluorophenyl)-3-hydroxymethyl-1-methyl-1,2,3,6tetrahydropyridine were dissolved in 350 ml of 99% ethanol, 5 g of 5% palladium on carbon were added, and the mixture was treated with hydrogen until 4500 ml were absorbed. The catalyst was filtered off, and the solution was evaporated to yield 37.5 g of (+)-b-4-(4-fluorophenyl)-3-hydroxymethyl1-methylpiperidine.
To a solution of sodium in methanol (125 ml) were added 3,4methylenedioxyphenol (29 g) and the (+)-b-4-(4-fluorophenyl)-3hydroxymethyl-1-methylpiperidine (37,5 g). The mixture was stirred and refluxed. After removal of the solvent in vacuo, the evaporation residue was poured into a mixture of ice (150 g), water (150 ml), and ether (200 ml). The ether layer was separated, and the aqueous layer was extracted with ether. The combined ether solutions were washed with water and dried with anhydrous magnesium sulphate, and the ether was evaporated. The residue was triturated with 200 ml of 99% ethanol and 11.5 ml of concentrated hydrochloric acid, yielding 30 g of (-)-b-4-(4-fluorophenyl-3-(1,3-benzdioxolyl(3)-oxymethyl)-1-methylpiperidine, hydrochloride were obtained. Melting point 202°C.
brand name
Paxil[as hydrochloride] (SmithKline Beecham);Seroxat.
Therapeutic Function
Antidepressant
Biological Functions
Paroxetine (Paxil) has an elimination half-life of 21 hours and is also highly bound to plasma proteins, so it requires special attention when administered with drugs such as warfarin. Paroxetine is a potent inhibitor of the cytochrome P450 2D6 isoenzyme and can raise the plasma levels of drugs metabolized via this route. Of particular concern are drugs with a narrow therapeutic index, such as TCAs and the type 1C antiarrhythmics flecainide, propafenone, and encainide. Additionally, paroxetine itself is metabolized by this enzyme and inhibits its own metabolism, leading to nonlinear kinetics. Weight gain is higher with paroxetine than with the other SSRIs, and it tends to be more sedating, presumably because of its potential anticholinergic effects. Additionally, patients have had difficulty with abrupt discontinuation with this agent, reporting a flulike syndrome; this symptom can be avoided by tapering the medication.
General Description
In the structure of paroxetine (Paxil), an amino group, protonatedin vivo could H-bond with the–CH2–O– unshared electrons.A β-arylamine–like structure with an extra aryl groupresults. The compound is a very highly selective SERT. Asexpected, it is an effective antidepressant and anxiolytic.
Pharmacokinetics
Paroxetine treats the symptoms of depression, various anxiety disorders, posttraumatic stress disorder, obsessive-compulsive disorder, and the vasomotor symptoms of menopause via the inhibition of serotonin reuptake. The onset of action of paroxetine is reported to be approximately 6 weeks.
Due its serotonergic activity, paroxetine, like other SSRI drugs, may potentiate serotonin syndrome. This risk is especially high when monoamine oxidase (MAO) inhibitors are given within 2 weeks of paroxetine administration. Upon cessation of MAO inhibitors, a 2-week interval before paroxetine administration is recommended. Do not coadminister these agents.
Pharmacokinetics
Paroxetine appears to be slowly but well absorbed from the GItract following oral administration with an oral
bioavailability of approximately 50%, suggesting first-pass metabolism, reaching peak plasma
concentrations in 2 to 8 hours. Food does not substantially affect the absorption of paroxetine. Paroxetine is
distributed into breast milk. Approximately 80% of an oral dose of paroxetine is oxidized by CYP2D6 to a
catechol intermediate, which is then either O-methylated or O-glucuronidated. These conjugates are then
eliminated in the urine.
Paroxetine exhibits a preincubation-dependent increase in inhibitory potency of CYP2D6 consistent with a
mechanism-based inhibition of CYP2D6. The inactivation of CYP2D6 occurs via the formation of an
o-quinonoid reactive metabolite.
The methylenedioxy has been associated with mechanism-based inactivation of other CYP isoforms.
In contrast, fluoxetine, a potent inhibitor of CYP2D6 activity, did not exhibit a mechanism-based inhibition of
CYP2D6. As a result of mechanism-based inhibition, saturation of CYP2D6 at clinical doses appears to
account for its nonlinear pharmacokinetics observed with increasing dose and duration of paroxetine
treatment, which results in increased plasma concentrations of paroxetine at low doses. The elderly may be
more susceptible to changes in doses and, therefore, should be started off at lower doses. Following oral
administration, paroxetine and its metabolites are excreted in both urine and feces.
Oral administration of a single dose resulted in unmetabolized paroxetine accounting for 2% and metabolites
accounting for 62% of the excretion products. The effect of age on the elimination of paroxetine suggests that
hepatic clearance of paroxetine can be reduced, leading to an increase in elimination half-life (e.g., to ~36
hours) and increased plasma concentrations. The metabolites of paroxetine have been shown to possess no
more than 2% of the potency of the parent compound as inhibitors of 5-HT reuptake; therefore, they are
essentially inactive.
Because paroxetine is a potent mechanism-based inhibitor of CYP2D6, this type of inhibition yields nonlinear
and long-term effects on drug pharmacokinetics, because the inactivated or complexed CYP2D6 must be
replaced by newly synthesized CYP2D6 protein. Thus, coadministration of paroxetine with CYP2D6-
metabolized medications should be closely monitored or, in certain cases, avoided, as should upward dose
adjustment of paroxetine itself.
Clinical Use
In vitro binding studies suggest that paroxetine is a more selective and potent inhibitor of 5-HT reuptake than fluoxetine. The drug essentially has no effect on NE or dopamine reuptake, nor does it show affinity for other neuroreceptors. Its onset of action is 1 to 4 weeks.
Veterinary Drugs and Treatments
Paroxetine may be beneficial for the treatment of canine aggression, and stereotypic or other obsessive-compulsive behaviors. It has been used occasionally in cats as well.
Drug interactions
Potentially hazardous interactions with other drugs
Analgesics: increased risk of bleeding with aspirin
and NSAIDs; risk of CNS toxicity increased with
tramadol; concentration of methadone possibly
increased.
Anti-arrhythmics: possibly inhibits propafenone
metabolism (increased risk of toxicity).
Anticoagulants: effect of coumarins possibly
enhanced; possibly increased risk of bleeding with
dabigatran.
Antidepressants: avoid concomitant use with MAOIs
and moclobemide (increased risk of toxicity); avoid
with St John’s wort; possibly enhanced serotonergic
effects with duloxetine; can increase concentration of
tricyclics; possible increased risk of convulsions with
vortioxetine.
Antiepileptics: antagonism (lowered convulsive
threshold); concentration reduced by phenytoin and
phenobarbital.
Antimalarials: avoid with artemether/lumefantrine
and piperaquine with artenimol.
Antipsychotics: concentration of clozapine and
possibly risperidone increased; metabolism
of perphenazine inhibited, reduce dose of
perphenazine; possibly inhibits aripiprazole
metabolism, reduce aripiprazole dose; concentration
possibly increased by asenapine; increased risk of
ventricular arrhythmias with pimozide - avoid.
Antivirals: concentration possibly reduced by
darunavir and ritonavir.
Beta blockers: concentration of metoprolol possibly
increased - increased risk of AV block - avoid in
cardiac insufficiency.
Dapoxetine: possible increased risk of serotonergic
effects - avoid.
Dopaminergics: increased risk of hypertension and
CNS excitation with selegiline - avoid; increased
risk of CNS toxicity with rasagiline - avoid.
Hormone antagonists: metabolism of tamoxifen to
active metabolite possibly reduced - avoid.
5HT1 agonists: risk of CNS toxicity increased by
sumatriptan - avoid; possibly increased risk of
serotonergic effects with naratriptan.
Lithium: increased risk of CNS effects - monitor
levels.
Methylthioninium: risk of CNS toxicity - avoid if
possible.
Metabolism
Paroxetine metabolism occurs in the liver and is largely mediated by cytochrome CYP2D6 with contributions from CYP3A4 and possibly other cytochrome enzymes. Genetic polymorphisms of the CYP2D6 enzyme may alter the pharmacokinetics of this drug. Poor metabolizers may demonstrate increased adverse effects while rapid metabolizers may experience decreased therapeutic effects.
The majority of a paroxetine dose is oxidized to a catechol metabolite that is subsequently converted to both glucuronide and sulfate metabolites via methylation and conjugation. In rat synaptosomes, the glucuronide and sulfate conjugates have been shown to thousands of times less potent than paroxetine itself. The metabolites of paroxetine are considered inactive.
Metabolism
Paroxetine is extensively metabolised in the liver to
pharmacologically inactive metabolites.
Urinary excretion of unchanged paroxetine is generally
less than 2% of dose whilst that of metabolites is about
64% of dose. About 36% of the dose is excreted in faeces,
probably via the bile, of which unchanged paroxetine
represents less than 1% of the dose. Thus paroxetine is
eliminated almost entirely by metabolism.
Properties of Paroxetine
| Melting point: | 114-116°C |
| Boiling point: | 451.7±45.0 °C(Predicted) |
| Density | 1.1844 (estimate) |
| storage temp. | under inert gas (nitrogen or Argon) at 2–8 °C |
| pka | pKa 9.51 (Uncertain) |
| form | Solid |
| color | Off-white to light yellow |
| CAS DataBase Reference | 61869-08-7(CAS DataBase Reference) |
| NIST Chemistry Reference | Paroxetine(61869-08-7) |
| EPA Substance Registry System | Paroxetine (61869-08-7) |
Safety information for Paroxetine
| Signal word | Warning |
| Pictogram(s) |
 Exclamation Mark Irritant GHS07 |
| GHS Hazard Statements |
H302:Acute toxicity,oral H315:Skin corrosion/irritation H319:Serious eye damage/eye irritation H335:Specific target organ toxicity, single exposure;Respiratory tract irritation |
| Precautionary Statement Codes |
P261:Avoid breathing dust/fume/gas/mist/vapours/spray. P305+P351+P338:IF IN EYES: Rinse cautiously with water for several minutes. Remove contact lenses, if present and easy to do. Continuerinsing. |
Computed Descriptors for Paroxetine
Paroxetine manufacturer
New Products
4,4-Difluoropiperidine hydrochloride tert-butyl 9-methoxy-3-azaspiro[5.5]undecane-3-carboxylate Indole Methyl Resin N-Isopropylurea N,N-Dicyclohexylcarbodiimide(DCC) MELDRUMS ACID 5-METHYLISOXAZOLE-4-CARBOXYLIC ACID Magnessium Bis glycinate Zinc ascorbate 1-bromo-2-butyne 2-acetamidophenol 9(10H)-anthracenone Erythrosin B, 4-Piperidinopiperidine 2-((4-morpholinophenylamino) (methylthio) methylene) malononitrile 2,4-dihydroxybenzaldehyde 3-(4-morpholinophenylamino)-5-amino-1H-pyrazole-4-carbonitrile Methyl 2-methylquinoline-6-carboxylate 2,6-dichloro-4-nitropyridine 4-Bromo-2-chlorobenzonitrile 2-(benzylamino)acetic acid hydrochloride 4-(tert-Butoxycarbonylamino)but- 2-ynoic acid 3,4-dihydro-2H-benzo[b][1,4]dioxepine 1-Phenyl-1-cycloprppanecarboxylicacidRelated products of tetrahydrofuran


![3S,4R-4-[phenyl-1-methylpiperidinyl] methanol](https://img.chemicalbook.in/CAS/20180906/GIF/176022-03-0.gif)
You may like
-
 61869-08-7 PAROXETINE 10.0% DC GRANULES 98%View Details
61869-08-7 PAROXETINE 10.0% DC GRANULES 98%View Details
61869-08-7 -
61869-08-7 98%View Details
61869-08-7 -
Paroxetine 98%View Details
61869-08-7 -
 61869-08-7 99%View Details
61869-08-7 99%View Details
61869-08-7 -
Paroxetine 97% CAS 61869-08-7View Details
61869-08-7 -
 Paroxetine 12 5mg TabletView Details
Paroxetine 12 5mg TabletView Details
61869-08-7 -
 Paroxetine 12 5mg TabletView Details
Paroxetine 12 5mg TabletView Details
61869-08-7 -
 40 mg Paroxetine Tablets IPView Details
40 mg Paroxetine Tablets IPView Details
61869-08-7