
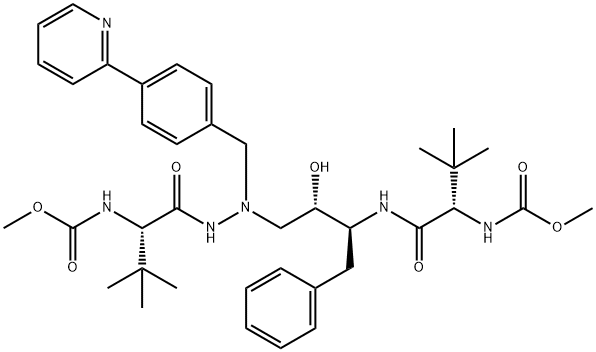
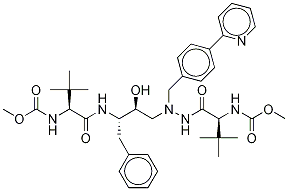
Atazanavir
Synonym(s):1,14-Dimethyl (3S,8S,9S,12S)-3,12-bis(1,1-dimethylethyl)-8-hydroxy-4,11-dioxo-9-(phenylmethyl)-6-[[4-(2-pyridinyl)phenyl]methyl]-2,5,6,10,13-pentaazatetradecanedioate;BMS-232632
- CAS NO.:198904-31-3
- Empirical Formula: C38H52N6O7
- Molecular Weight: 704.86
- MDL number: MFCD08435966
- EINECS: 812-543-8
- SAFETY DATA SHEET (SDS)
- Update Date: 2025-12-17 11:34:44
What is Atazanavir?
Absorption
Atazanavir is rapidly absorbed with a Tmax of approximately 2.5 hours. Atazanavir demonstrates nonlinear pharmacokinetics with greater than dose-proportional increases in AUC and Cmax values over the dose range of 200 to 800 mg once daily. A steady state is achieved between Days 4 and 8, with an accumulation of approximately 2.3-fold.
Administration of atazanavir with food enhances bioavailability and reduces pharmacokinetic variability. Administration of a single 400-mg dose of atazanavir with a light meal (357 kcal, 8.2 g fat, 10.6 g protein) resulted in a 70% increase in AUC and 57% increase in Cmax relative to the fasting state. Administration of a single 400-mg dose of atazanavir with a high-fat meal (721 kcal, 37.3 g fat, 29.4 g protein) resulted in a mean increase in AUC of 35% with no change in Cmax relative to the fasting state. Administration of atazanavir with either a light or high-fat meal decreased the coefficient of variation of AUC and Cmax by approximately one-half compared to the fasting state.
Coadministration of a single 300-mg dose of atazanavir and a 100-mg dose of ritonavir with a light meal (336 kcal, 5.1 g fat, 9.3 g protein) resulted in a 33% increase in the AUC and a 40% increase in both the Cmax and the 24-hour concentration of atazanavir relative to the fasting state. Coadministration with a high-fat meal (951 kcal, 54.7 g fat, 35.9 g protein) did not affect the AUC of atazanavir relative to fasting conditions and the Cmax was within 11% of fasting values. The 24-hour concentration following a high-fat meal was increased by approximately 33% due to delayed absorption; the median Tmax increased from 2.0 to 5.0 hours. Coadministration of atazanavir with ritonavir with either a light or a high-fat meal decreased the coefficient of variation of AUC and Cmax by approximately 25% compared to the fasting state.
Description
Atazanavir is an inhibitor of human immunodeficiency virus type 1 (HIV-1) protease, an enzyme that is essential for the processing of Gag and Gag-Pol polyproteins into structural and enzymatic proteins required for viral replication. It has a similar pharmacophore motif to the other six widely marketed HIV protease inhibitors, most of which are based upon a hydroxyethylamine template. Uniquely, it possesses an aza-peptide motif but maintains many similar pharmacophore elements including lipophilic moieties that presumably bind to S2, S1, S′1 , and S′2 positions. Atazanavir is pseudo-symmetric about the central template, incorporating D-tert-Leucine at both termini. This compound is synthesized in about seven steps, with a key coupling of the chiral epoxide (derived from phenylalanine and imparting one chiral center) and N-tert-boc-N′-(4-[2-pyridyl]benzyl)hydrazine. Removal of both tert-Boc groups and double acylation with methoxycarbonyl-tert-Leucine provides the product. Another synthesis of atazanavir entails ten steps and utilizes α-(tert-bocamino) phenylpropanal as a chiral intermediate. It is a potent inhibitor of indinavir-resistant and saquinavir-resistant strains of HIV-1 (IC50=0.03–0.1 and 0.04–0.1 μM, respectively). In 300 patients who had failed previous treatment, atazanavir (400 mg once daily) was compared to lopinavir (400 mg twice daily) and ritonavir (100 mg); both arms additionally receiving two non-reverse transcriptase inhibitors. After 24 weeks, HIV RNA levels of <400 copies/mL were noted in 61% of patients receiving atazanavir and 81% of those taking lopinavir/ritonavir. After 96 weeks of therapy with atazanavir, HIV RNA copy levels were found to be <400 and <50 in 80 and 58% of patients, respectively. A study of the cross-resistance profile relative to other protease inhibitors using a panel of 551 clinical isolates (without prior atazanavir exposure but with cross-resistance to one or two other protease inhibitors; the majority had resistance to nelfinavir) showed that greater than 80% retained susceptibility to atazanavir. All of the resistant isolates from patients taking atazanavir had an I50 L substitution. The recommended dosage of atazanavir is 400 mg once daily. It has a mean half-life range of 7.9–6.5 h with about 60% bioavailability and moderate plasma protein binding (86% albumin and 89% alpha-1- acid glycoprotein (AAG)). Atazanavir was well tolerated in clinical studies and it displayed minimal lipid modulation when tested in combination with two non-reverse transcriptase inhibitors. Atazanavir had no effect on total cholesterol, low-density lipoprotein, and triglyceride levels when compared with other protease inhibitors that caused sustained elevations in these lipid levels.
Chemical properties
Crystalline Solid
Originator
Novartis (US)
The Uses of Atazanavir
Atazanavir is a novel azapeptide protease inhibitor (PI)
The Uses of Atazanavir
enzyme inhibitor
The Uses of Atazanavir
Atazanavir is a novel azapeptide HIV protease inhibitor (PI). Antiviral.
The Uses of Atazanavir
Atazanavir is an inhibitor of HIV-1 protease (EC50 = 2.6 nM). In isolated cells, it has additive to moderately synergistic antiviral effects when combined with other antiretroviral drugs. As a result, it is commonly used in vivo in combination therapy for HIV-1 infection. Atazanavir competitively inhibits UDP-gluronosyltransferase, which conjugates bilirubin for clearance, leading to hyperbilirubinemia in a significant portion of those receiving atazanavir therapy.
What are the applications of Application
Atazanavir is a highly potent HIV-1 protease inhibitor
Background
Atazanavir (formerly known as BMS-232632) is an antiretroviral drug of the protease inhibitor (PI) class. Like other antiretrovirals, it is used to treat infection of human immunodeficiency virus (HIV). Atazanavir is distinguished from other PIs in that it can be given once daily (rather than requiring multiple doses per day) and has lesser effects on the patient's lipid profile (the amounts of cholesterol and other fatty substances in the blood). Like other protease inhibitors, it is used only in combination with other HIV medications. The U.S. Food and Drug Administration (FDA) approved atazanavir on June 20, 2003.
Indications
Atazanavir is indicated in combination with other antiretroviral agents for the treatment of HIV-1 infection in adults and pediatric patients 3 months of age and older weighing at least 5kg. Atazanavir is also indicated in combination with cobicistat and other antiretrovirals for the treatment of HIV-1 infection in adults and pediatric patients weighing at least 35kg.
Definition
ChEBI: A heavily substituted carbohydrazide that is an antiretroviral drug of the protease inhibitor (PI) class used to treat infection of human immunodeficiency virus (HIV).
brand name
Reyataz (Bristol-Myers Squibb).
Acquired resistance
Mutations at positions 50 (I50L), 84 (I84V) and 88 (N88S) of the protease gene are associated with resistance.
General Description
Atazanavir is an antiretroviral agent that has been approvedby the FDA for use in combination with other anti-RTagents for the treatment of HIV infections. The drug is alwaysused in combination with RT inhibitors.
Pharmaceutical Applications
An azapeptide formulated as the sulfate for oral use.
Biochem/physiol Actions
Atazanavir is an antiviral HIV protease inhibitor.
Mechanism of action
Atazanavir is dosed orally once daily, thus reducing "pill burden," and it appears to have minimal impact on lipid parameters but does increase total bilirubin. The drug is well absorbed when administered orally with food (bioavailability, ~68%). The drug is highly bound to plasma protein (86%) and is metabolized by CYP3A isoenzyme. Atazanavir is a moderate inhibitor of CYP3A, and potential drug–drug interactions are possible with CYP3A inhibitors and inducers.
Pharmacokinetics
Atazanavir (ATV) is an azapeptide HIV-1 protease inhibitor (PI) with activity against Human Immunodeficiency Virus Type 1 (HIV-1). HIV-1 protease is an enzyme required for the proteolytic cleavage of the viral polyprotein precursors into the individual functional proteins found in infectious HIV-1. Atazanavir binds to the protease active site and inhibits the activity of the enzyme. This inhibition prevents cleavage of the viral polyproteins resulting in the formation of immature non-infectious viral particles. Protease inhibitors are almost always used in combination with at least two other anti-HIV drugs. Atazanivir is pharmacologically related but structurally different from other protease inhibitors and other currently available antiretrovirals.
Atazanavir exhibits anti-HIV-1 activity with a mean 50% effective concentration (EC50) in the absence of human serum of 2 to 5 nM against a variety of laboratory and clinical HIV-1 isolates grown in peripheral blood mononuclear cells, macrophages, CEM-SS cells, and MT-2 cells.
Atazanavir has activity against HIV-1 Group M subtype viruses A, B, C, D, AE, AG, F, G, and J isolates in cell culture. Atazanavir has variable activity against HIV-2 isolates (1.9-32 nM), with EC50 values above the EC50 values of failure isolates. Two-drug combination antiviral activity studies with atazanavir showed no antagonism in cell culture with PIs (amprenavir, indinavir, lopinavir, nelfinavir, ritonavir, and saquinavir), NNRTIs (delavirdine, efavirenz, and nevirapine), NRTIs (abacavir, didanosine, emtricitabine, lamivudine, stavudine, tenofovir DF, and zidovudine), the HIV-1 fusion inhibitor enfuvirtide, and two compounds used in the treatment of viral hepatitis, adefovir and ribavirin, without enhanced cytotoxicity.
HIV-1 isolates with a decreased susceptibility to atazanavir have been selected in cell culture and obtained from patients treated with atazanavir or atazanavir with ritonavir. HIV-1 isolates with 93- to 183-fold reduced susceptibility to atazanavir from three different viral strains were selected in cell culture for 5 months. The substitutions in these HIV-1 viruses that contributed to atazanavir resistance include I50L, N88S, I84V, A71V, and M46I. Changes were also observed at the protease cleavage sites following drug selection. Recombinant viruses containing the I50L substitution without other major PI substitutions were growth impaired and displayed increased susceptibility in cell culture to other PIs (amprenavir, indinavir, lopinavir, nelfinavir, ritonavir, and saquinavir). The I50L and I50V substitutions yielded selective resistance to atazanavir and amprenavir, respectively, and did not appear to be cross-resistant.
Concentration- and dose-dependent prolongation of the PR interval in the electrocardiogram has been observed in healthy subjects receiving atazanavir. In placebo-controlled Study AI424-076, the mean (±SD) maximum change in PR interval from the predose value was 24 (±15) msec following oral dosing with 400 mg of atazanavir (n=65) compared to 13 (±11) msec following dosing with placebo (n=67). The PR interval prolongations in this study were asymptomatic. There is limited information on the potential for a pharmacodynamic interaction in humans between atazanavir and other drugs that prolong the PR interval of the electrocardiogram.
Electrocardiographic effects of atazanavir were determined in a clinical pharmacology study of 72 healthy subjects. Oral doses of 400 mg (maximum recommended dosage) and 800 mg (twice the maximum recommended dosage) were compared with placebo; there was no concentration-dependent effect of atazanavir on the QTc interval (using Fridericia’s correction). In 1793 subjects with HIV-1 infection, receiving antiretroviral regimens, QTc prolongation was comparable in the atazanavir and comparator regimens. No atazanavir-treated healthy subject or subject with HIV-1 infection in clinical trials had a QTc interval >500 msec
Pharmacokinetics
Oral absorption: c. 68%
Cmax 400 mg once daily: c. 3.15 μg/L
300 mg + ritonavir 100 mg once daily: c. 4.47 μg/L
Cmin 400 mg once daily: c. 0.27 μg/L
300 mg + ritonavir 100 mg once daily: c. 0.65 μg/L
Plasma half-life: c. 8.6 h (300 mg+ ritonavir
100 mg)
Volume of distribution: c. Not known/available
Plasma protein binding: c. 86%
Absorption
Administration with food enhances bioavailability and reduces pharmacokinetic variability. Absorption is dependent on gastric pH. It should be given separately from proton-pump inhibitors or H2-receptor antagonists. Buffered or entericcoated formulations should be given (with food) 2 h before or 1 h after co-administration of didanosine.
Distribution
It penetrates moderately well into the CNS. The semen:plasma ratio is 0.11–4.42. It is distributed into breast milk.
Metabolism
It is extensively metabolized by CYP3A4. Administration with ritonavir prevents metabolization and enhances the pharmacokinetic profile.
Excretion
Following a single 400 mg dose, 79% and 13% of the dose was recovered in the feces and urine, respectively. It should be used with caution in the presence of mild hepatic impairment and should not be used in patients with more severe hepatic impairment.
Clinical Use
Treatment of HIV infection (in combination with other antiretroviral drugs)
Side Effects
The most common adverse reactions (≥2%) are nausea, jaundice/ scleral icterus, rash, headache, abdominal pain, vomiting, insomnia, peripheral neurological symptoms, dizziness, myalgia, diarrhea, depression and fever.
Drug interactions
Potentially hazardous interactions with other drugs
Anti-arrhythmics: possibly increased levels of
amiodarone and lidocaine.
Antibacterials: concentration of both drugs
increased when given with clarithromycin; rifabutin
concentration increased - reduce dose of rifabutin;
rifampicin reduces atazanavir concentration - avoid;
avoid with telithromycin in severe renal and hepatic
impairment.
Anticoagulants: avoid with apixaban and
rivaroxaban.
Antidepressants: concentration reduced by St John’s
wort - avoid.
Antifungals: concentration increased by
posaconazole; concentration of voriconazole
increased or decreased, concentration of atazanavir
also reduced.
Antimalarials: avoid with artemether/lumefantrine;
may increase quinine concentration.
Antipsychotics: possibly inhibits metabolism of
aripiprazole - reduce dose of aripiprazole; possibly
increased concentration of pimozide and quetiapine
- avoid.
Antivirals: concentration reduced by boceprevir;
concentration of daclatasvir increased, reduce dose
of daclatasvir; absorption reduced by didanosine
tablets; concentration reduced by efavirenz -
avoid; concentration of elvitegravir increased
when atazanavir boosted with ritonavir - reduce
elvitegravir dose; concentration possibly reduced by
nevirapine - avoid; concentration of paritaprevir
increased; increased risk of ventricular arrhythmias
with saquinavir - avoid; concentration reduced
by tenofovir and tenofovir concentration possibly
increased; avoid with indinavir; concentration
of maraviroc increased, consider reducing
dose of maraviroc; possibly reduces telaprevir
concentration, also concentration of atazanavir
increased; concentration of tipranavir increased,
also concentration of atazanavir reduced; avoid
with elbasvir/grazoprevir, increased grazoprevir
concentration.
Anxiolytics and hypnotics: possibly increases
concentration of midazolam - avoid with oral
midazolam.
Calcium-channel blockers: concentration of
diltiazem increased - reduce dose of diltiazem;
possibly increased verapamil concentration.
Ciclosporin: possibly increased concentration of
ciclosporin.
Colchicine: possibly increases risk of colchicine
toxicity, avoid in hepatic or renal impairment.
Cytotoxics: possibly increases concentration of
axitinib, reduce dose of axitinib; possibly increases
concentration of bosutinib, avoid or reduce dose;
possibly increases concentration of crizotinib and
everolimus - avoid; avoid with cabazitaxel and
pazopanib; concentration of ibrutinib possibly
increased, reduce dose of ibrutinib; possibly inhibits
metabolism of irinotecan - increased risk of toxicity.
Dapoxetine: avoid concomitant use, increased risk of
toxicity.
Ergot alkaloids: possibly increased concentration of
ergot alkaloids - avoid.
Orlistat: absorption possibly reduced by orlistat.
Ranolazine: possibly increases ranolazine
concentration - avoid.
Sildenafil: possibly increased side effects of sildenafil.
Sirolimus: possibly increased concentration of
sirolimus.
Statins: avoid with simvastatin - increased risk of
myopathy; possibly increased risk of myopathy with atorvastatin, pravastatin and rosuvastatin - reduce
rosuvastatin dose.
Tacrolimus: possibly increased concentration of
tacrolimus.
Ticagrelor: possibly increases concentration of
ticagrelor - avoid.
Ulcer-healing drugs: concentration significantly
reduced by omeprazole and esomeprazole and
possibly other proton pump inhibitors - avoid;
concentration possibly reduced by histamine H2
antagonists.
Metabolism
Atazanavir is extensively metabolized in humans. The major biotransformation pathways of atazanavir in humans consisted of monooxygenation and dioxygenation. Other minor biotransformation pathways for atazanavir or its metabolites consisted of glucuronidation, N-dealkylation, hydrolysis, and oxygenation with dehydrogenation. Two minor metabolites of atazanavir in plasma have been characterized. Neither metabolite demonstrated in vitro antiviral activity. In vitro studies using human liver microsomes suggested that atazanavir is metabolized by CYP3A.
Metabolism
Atazanavir is principally metabolised by CYP3A4
isozyme to oxygenated metabolites. Metabolites are
then excreted in the bile as either free or glucuronidated
metabolites. Additional minor metabolic pathways consist
of N-dealkylation and hydrolysis. Two minor metabolites
of atazanavir in plasma have been characterised. Neither
metabolite demonstrated in vitro antiviral activity.
Following a single 400 mg dose of [14C]-atazanavir, 79%
and 13% of the total radioactivity was recovered in the
faeces and urine, respectively. Unchanged drug accounted
for approximately 20% and 7% of the administered dose
in the faeces and urine, respectively.
Properties of Atazanavir
| Melting point: | 207-2090C |
| alpha | D -47° (c = 1 in ethanol) |
| Density | 1.178±0.06 g/cm3(Predicted) |
| storage temp. | -20°C |
| solubility | Ethanol (Slightly), Methanol (Slightly) |
| form | powder |
| pka | 11.11±0.46(Predicted) |
| color | white to beige |
| CAS DataBase Reference | 198904-31-3 |
Safety information for Atazanavir
| Signal word | Warning |
| Pictogram(s) |
 Exclamation Mark Irritant GHS07 |
| GHS Hazard Statements |
H319:Serious eye damage/eye irritation |
| Precautionary Statement Codes |
P305+P351+P338:IF IN EYES: Rinse cautiously with water for several minutes. Remove contact lenses, if present and easy to do. Continuerinsing. |
Computed Descriptors for Atazanavir
Atazanavir manufacturer
New Products
Indole Methyl Resin tert-butyl 9-methoxy-3-azaspiro[5.5]undecane-3-carboxylate Boc-His(Boc)-OH 2-CTC Resin 4-Chloro-7-tosy1-7Hpyrrolo[2,3-d]pyrimidine 5,7-Dibromo-1H-indole 2,5-dichloro-N-hydroxy-4,6-dimethylpyridine-3-carboximidamide 2,2-Dimethoxy-7-azaspiro[3.5]nonane hydrochloride 4-chloromethyl-5-methyl-1,3-dioxol-2-one (DMDO-Cl) R-2-BENZYLOXY PROPIONIC ACID 1,1’-CARBONYLDIIMIDAZOLE 1,1’-CARBONYLDI (1,2-4 TRIAZOLE) N-METHYL INDAZOLE-3-CARBOXYLIC ACID 4-((2-hydroxyethyl)thio)benzoic acid 1-(TERT-BUTOXYCARBONYL)-2-PYRROLIDINONE Methyl 6-methylnicotinate 3-Pyridineacrylic acid tert-Butyl carbazate TETRAHYDRO-2H-PYRAN-3-OL 2-((4-morpholinophenylamino) (methylthio) methylene) malononitrile 3-(4-morpholinophenylamino)-5-amino-1H-pyrazole-4-carbonitrile 2,4-dihydroxybenzaldehyde 1,3-Diethyl-1,3-Diphenylurea Methyl 2-methylquinoline-6-carboxylateRelated products of tetrahydrofuran

You may like
-
 Atazanavir 98%View Details
Atazanavir 98%View Details -
 Atazanavir 98%View Details
Atazanavir 98%View Details -
 Atazanavir 99%View Details
Atazanavir 99%View Details -
 Atazanavir 98%View Details
Atazanavir 98%View Details
198904-31-3 -
 Atazanavir 198904-31-3 98%View Details
Atazanavir 198904-31-3 98%View Details
198904-31-3 -
 Atazanavir 98% (HPLC) CAS 198904-31-3View Details
Atazanavir 98% (HPLC) CAS 198904-31-3View Details
198904-31-3 -
Atazanavir for system suitability CAS 198904-31-3View Details
198904-31-3 -
Atazanavir CAS 198904-31-3View Details
198904-31-3