

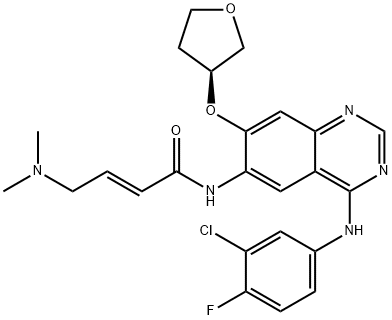
Afatinib
| Price | USD7.00 |
| Packge | 1KG |
- Min. Order:1KG
- Supply Ability:JD 485
- Time:2019-09-02
Product Details
- Product Name Afatinib
- CAS No. 850140-72-6
- MFC24H25ClFN5O3
- MW485.94
- AppearanceYellow powder.Pale yellow
- density 1.380
- Boiling point 676.9±55.0 °C(Predicted)
- Melting point 102 °C
- storage temp. -20°C
▼
▲
Product Name:
Synonyms:
(2E)-N-[4-[(3-Chloro-4-fluorophenyl)amino]-7-[[(3S)-tetrahydro-3-furanyl]oxy]-6-quinazolinyl]-4-(dimethylamino)-2-butenamide;4-[(3-chloro-4-fluorophenyl)aMino]-6-{[4-(N,N-diMethylaMino)-1-oxo-2-buten-1-yl]aMino}-7-((S)-tetrahydrofuran-3-yloxy)-quinazoline;2-ButenaMide, N-[4-[(3-chloro-4-fluorophenyl)aMino]-7-[[(3S)-tetrahydro-3-furanyl]oxy]-6-quinazolinyl]-4-(diMethylaMino)-, (2E)-;(S,E)-N-(4-((3-Chloro-4-fluorophenyl)amino)-7-((tetrahydrofuran-3-yl)oxy)quinazolin-6-yl)-4-(dime;Afatinib API;Afatinib(BIBW2992MA2);(2E)-N-{4-[(3-Chloro-4-fluorophenyl)amino]-7-[(3S)-oxolan-3-yloxy]quinazolin-6-yl}-4-(dimethylamino)but-2-enamide;Afatinib
CAS:
MF:
C24H25ClFN5O3
MW:
485.9384032
EINECS:
Product Categories:
Mol File:
▼
▲
Afatinib Chemical Properties
▼
▲
density
1.380
form
Yellow powder.
▼
▲
Safety Information
▼
▲
HS Code
29420000
▼
▲
Afatinib Usage And Synthesis
▼
▲
Afatinib is a prescription medicine that can be taken orally for the treatment of non-small cell lung cancer (NSCLC) with atypical Epidermal Growth Factor Receptor (EFGR) agents. Afatinib is also prescribed for the treatment of NSCLC with the potential or radiating to other body tissues other than the lungs (metastatic cancer) as detected through an FDA approved examination. Afatinib is a 4-anilinoquinazoline tyrosine kinase suppressor that is available as a dimaleate salt (brand name: Giltrif). The drug is the first FDA-endorsed oncology product by Boehringer Ingelheim.
The drug is a kinase agonist that is indicated as monotherapy for EFGR and tyrosine kinase suppressor-naive adult patients whose NSCLC is metastatic or locally advanced and the tumours are resistant to EGFR mutations as approved by an FDA-endorsed examination. In addition, the adult patients should indicate signs of squamous histology progress at the time of platinum-based chemotherapy or after the sessions.
The recommended dose of Afatinib is 40mg administered orally once every day 1-2 hours after the consumption of a meal, until the tumours regress or if the patient’s body can no longer tolerate the drug.
Afatinib is an irreversible ErbB inhibitor that forms covalent bonds with the kinase domains of EGFR, HER4, and human EGFRs (HER) 2, which results in an irreparable obstruction of tyrosine kinase autophosphorylation.
Afatinib as a single treatment agent obstructs the ErbB receptor which influences tumour regression and also inhibits further growth of the tumour through deregulation of the ErbB pathway. NSCLC tumours with a characteristic activating EGFR variation (Del 19, L858R) and other uncommon variations in exon 21 (L861Q) and exon 18 (G719X) are responsive to treatment with Afatinib in both clinical and non-clinical setups. Limited clinical and non-clinical activity indicates NSCLC tumours with positioning mutations in exon 20.
The accession of a secondary T790M variation is a crucial trait of acquired resistance to the drug and gene dosage of T790M-possessing allele correlates with partial resistance in vitro. The T790M variation is highlighted in about 50% of the patient’s tumour’s whose development on Afatinib may be perceived as a next-line therapeutic option where the T790M aims at EGFR TKIs. Other mechanisms of resistance to the drug have been highlighted MET gene extension clinically and preclinically.
The impact of multiple doses of the drug (50 mg once per day) on the QTc interval and the cardiac electrophysiology was examined in a single-arm, open-label study in people with refractory or relapsed solid tumours. The study maintains that there were no developments in the mean QTc interval, >20 ms.
Afatinib as a single treatment agent obstructs the ErbB receptor which influences tumour regression and also inhibits further growth of the tumour through deregulation of the ErbB pathway. NSCLC tumours with a characteristic activating EGFR variation (Del 19, L858R) and other uncommon variations in exon 21 (L861Q) and exon 18 (G719X) are responsive to treatment with Afatinib in both clinical and non-clinical setups. Limited clinical and non-clinical activity indicates NSCLC tumours with positioning mutations in exon 20.
The accession of a secondary T790M variation is a crucial trait of acquired resistance to the drug and gene dosage of T790M-possessing allele correlates with partial resistance in vitro. The T790M variation is highlighted in about 50% of the patient’s tumour’s whose development on Afatinib may be perceived as a next-line therapeutic option where the T790M aims at EGFR TKIs. Other mechanisms of resistance to the drug have been highlighted MET gene extension clinically and preclinically.
The impact of multiple doses of the drug (50 mg once per day) on the QTc interval and the cardiac electrophysiology was examined in a single-arm, open-label study in people with refractory or relapsed solid tumours. The study maintains that there were no developments in the mean QTc interval, >20 ms.
An oral administration of Afatinib attains peak plasma concentrations after 2-5 hours. The geometric mean relative bioavailability of 20mg tablets is 92% as opposed to the oral syrup. Systemic exposure to the drug is reduced by 50% and 30%, Cmax and AUC0-∞ respectively, when taken with a high-fat meal as opposed to its administration after a period of fasting. The population pharmacokinetic data obtained from several clinical trials on tumours suggests that there is an average reduction of 26% in AUCss if a patient consumes food about 1-3 hours prior to the administration of Afatinib.
The volume of distribution of the drug in healthy volunteers is 4500 L. The high volume of transmission in plasma indicates that there could be a higher rate of distribution in body tissues.
Metabolic reactions that are catalyzed by enzymes play an insignificant role in the synthesis of Afatinib. Covalent bonds to proteins are the fundamental forms of the drug’s metabolites.
Afatinib may interact with phenobarbital, St. Johns wort, phenytoin, carbamazepine, rifampicin, saquinavir, nelfinavir, tacrolimus, quinidine, itraconazole, amiodarone, verapamil, ketoconazole, cyclosporine A. or erythromycin. This is not an all-inclusive list of all the drugs that may interact with Afatinib hence a patient should always notify their healthcare provider of any herbal supplements, over-the-counter drugs and other medications they are taking prior to the initiation of treatment.
Afatinib may also harm a developing fetus hence it is not recommended during pregnancy.
Afatinib may also harm a developing fetus hence it is not recommended during pregnancy.
The elimination of Afatinib in humans is primarily through the faecal route. The administration of an oral solution of the drug suggests that about 85.4 % of Afatinib is recovered in faeces whereas 4.3% of the drug in urine.
Common side effects associated with the administration of Afatinib include conjunctivitis, fever, runny nose, bloody nose, bladder/urinary tract infection, itching, vomiting, weight loss, loss of appetite nausea, acne, dry skin, skin infections around toenails or fingernails, mouth sores, chapped lips, inflammation of the lips and the mouth, blisters or skin lesions, skin rash, and diarrhoea.
Adverse reactions to Afatinib may include Keratitis, hepatic toxicity, Interstitial Lung Disease, Exfoliative and Bullous Skin Disorders, and diarrhoea.
Adverse reactions to Afatinib may include Keratitis, hepatic toxicity, Interstitial Lung Disease, Exfoliative and Bullous Skin Disorders, and diarrhoea.
Uses
An aminocrotonylamino-substituted quinazoline derivative
Definition
ChEBI: A quinazoline compound having a 3-chloro-4-fluoroanilino group at the 4-position, a 4-dimethylamino-trans-but-2-enamido group at the 6-position, and an (S)-tetrahydrofuran-3-yloxy group at the 7-position. Used (as its di aleate salt) for the first-line treatment of patients with metastatic non-small cell lung cancer.
Indications
The collection of ibrutinib (Imbruvica(R), Pharmacyclics Inc.), afatinib, and osimertinib represents the small, yet expanding, group of covalent SMKIs. Ibrutinib is a non-receptor Bruton’s tyrosine kinase inhibitor approved for the treatment of relapsed chronic lymphocytic leukemia. Afatinib, approved for NSCLC in 2013 and squamous NSCLC in 2016, is a second-generation irreversible EGFR inhibitor that targets wild-type EGFR, the mutant T790M EGFR, and HER2. Osimertinib (AZD9291), which was approved by FDA in November 2015, is a third-generation irreversible EGFR inhibitor that selectively targets the mutant T790M EGFR. Rociletinib, which shares a high degree of structural similarity with that of osimertinib, is a promising covalent EGFR inhibitor developed by Clovis Oncology aimed for the treatment of patients with EGFR T790M-mutated NSCLC, until the company terminated its development in May 2016 following a negative vote fromthe FDA’sOncologic Drugs Advisory Committee.
Biological Activity
Afatinib is an irreversible kinase inhibitor and binds to the kinase domains of EGFR (ErbB1), HER2 (ErbB2), and HER4 (ErbB4) to inhibit tyrosine kinase autophosphorylation. This results in a downregulation of ErbB signaling and subsequent inhibition of proliferation of cell lines expressing wild-type EGFR, selected EGFR exon 19 deletion mutations, or exon 21 L858R mutations. It also inhibited in vitro proliferation of cell lines overexpressing HER2. Overall, tumor growth was inhibited by Afatinib very effectively with low nanomolar IC50 values ranging from approximately 6nM to below 500 nM.
Enzyme inhibitor
This oral quinazoline derivative and EGFR/HER2-directed protein kinase inhibitor (FW = 485.94; CASs = 439081-18-2 (free base), 936631-70-8 (maleic acid salt), 1254955-21-9 (HCl salt); Solubility (at 25°C): 197 mg/mL DMSO, 1 mg/mL Water), also known by its code name BIBW2992, its trade names Gilotrif? Tomtovok?, Tovok?, and its systematic name (S,E)-N-(4-(3-chloro-4-fluorophenyl-amino)-7-(tetrahydrofuran- 3-yloxy)quinazolin-6-yl)-4-(dimethylamino)but-2-enamide, irreversibly inactivates EGFRwt, EGFRL858R, EGFRL858R/T790M and HER2with IC50 values of 0.5 nM, 0.4 nM, 10 nM and 14 nM, respectively. Mode of Action: The irreversible binding of afatinib to HER2 inactivates its interactions with a preferred partner of EGFR, and blocking the HER2- EGFR heterodimerization reduces their intrinsic tyrosine kinase activities. Irreversible inhibitors (such as afatinib and dacomitinib) that target all ErbB family receptor tyrosine kinases are intended to confer sustained disease control in ErbB-dependent cancers. Because nearly all EGFRmutated patients eventually develop resistance to reversible EGFR-TKIs after a median of 14 months, tafatinib’s irreversible action is thought to be a promising feature of its mode of action. Pharmacokinetics: Afatinib’s PK profile is best described by a two-compartment disposition model, with first-order absorption and linear elimination. There was a slightly more than proportional increase in exposure with increasing dose, most likely due to dose-dependent relative bioavailability. For the therapeutic dose of 40 mg, the estimated apparent total clearance rate at steady state was 734 mL/min.
Company Profile Introduction
Henan CoreyChem Co., Ltd, based on the original Zhengzhou Cote Chemical Research Institute, be brave in absorbing highly educated talents & overseas returnees; actively responded to Zhengzhou City High-tech Zone Government’s Special Care Policy, reorganized and founded in National University of Science and Technology Park, which is a high-tech, stock enterprise of high-end chemical Custom synthesis;The park was created by the People's Government of Henan Province, and proved by Ministry of Education and the National Science & Technology, taking the construction mode of "many college a park, and common development", mainly depends on Zhengzhou University and Henan University’s scientific research and talent advantage to set up Universities, scientific research institute and enterprise scientific research achievements transformation platform, to make high-tech enterprises incubate, is the new high-tech talent gathering base, high and new technology industry enterprise radiation base, colleges and universities technological innovation base.
Henan Coreychem Co., Ltd, facing global High-tech pharmaceutical raw materials, high complex new type intermediates, fine chemicals custom synthesis, scale-up production and Rare chemicals trade. Corey have well-equipped machine, strong technical force and considerate marketing team service. We also have rich experience advantage in basic research, small scale process development, scale-up, industrial technology development & production and cost control.
Recommended supplier
-
VIP1年
- Myosynth Laboratories Pvt Ltd
- 850140-72-6 Afatinib 98%
- Inquiry
- 2024-03-14
-
VIP1年
- Shreyaa Medilife Private Limited
- Afatinib 98%
- Inquiry
- 2024-03-04
-
VIP1年
- Sakar Healthcare
- 850140-72-6 98%
- Inquiry
- 2024-02-05
-
VIP1年
- Sai Life Sciences Ltd
- 850140-72-6 Afatinib 98%
- Inquiry
- 2024-01-23
-
VIP1年
- Alembic Pharmaceuticals Limited
- Afatinib 850140-72-6 98%
- Inquiry
- 2024-01-06
-
VIP1年
- Lupin Ltd
- Afatinib 99%
- Inquiry
- 2024-01-05
-
VIP2年
- AVD pharmaceuticals Pvt Ltd
- 850140-72-6 98%
- Inquiry
- 2023-09-11

- Since:2014-12-17
- Address: No.967,15th Floor,Unit 7, Building 1, No.70 of DianChang Road, High-tech Development Zone, Zhengzho
INQUIRY